
Manuscript accepted on :02-09-2022
Published online on: 18-10-2022
Plagiarism Check: Yes
Reviewed by: Dr. Amarendranath Choudhury
Second Review by: Dr. Mohammad Reza Mohammadabadi
Final Approval by: Dr. Josphert Ngui Kimatu
Sabritee Patel and Poonam Sharma*
and Poonam Sharma*
Department of Zoology, Gargi College, University of Delhi, Delhi, India.
Corresponding Author E-mail: poonam.sharma@gargi.du.ac.in
DOI : https://dx.doi.org/10.13005/bpj/2530
Abstract
Autism spectrum disorder (ASD) is a chronic neurological and developmental disability characterised by inability to develop social relationships, trouble expressing feelings, and repeated behaviours - clinically defined as stereotyped behaviour - that affect how people interact, learn, and behave. Because of the vast range of types and severity of symptoms, it is classified as a "spectrum" disorder. Over the last two decades, the prevalence of ASD has progressively increased, and one out of every 160 children worldwide is estimated to have an ASD. Over 75 percent of ASD patients show psychiatric disorders like depression, stress, bipolar disorder, Tourette syndrome, attention deficit hyperactivity disorder (ADHD). In the present study, in silico analysis was done to identify different rare mutations in genes implicated in ASD. Single nucleotide polymorphisms in ADNP, ARID1B, ASH1L, CHD2, CHD8, DYRK1A, POGZ, SHANK3, and SYNGAP1 genes were identified to be associated with ASD aetiology. A single mutation in these genes can result in defective chromatin remodeling, altering the function of several genes and potentially causing intellectual impairment and autism spectrum disorder (ASD). Understanding and analyzing these SNPs linked to ASD as risk factors can aid in the early detection and diagnosis of the disorder.
Keywords
Autism spectrum disorder; Chromatin remodeling; Histone methylation; Mutation; SNPs
Download this article as:| Copy the following to cite this article: Patel S, Sharma P. In silico Analysis of Common Autism Spectrum Disorder Genetic Risk Variations. Biomed Pharmacol J 2022;15(4). |
| Copy the following to cite this URL: Patel S, Sharma P. In silico Analysis of Common Autism Spectrum Disorder Genetic Risk Variations. Biomed Pharmacol J 2022;15(4). Available from: https://bit.ly/3s5IEW6 |
Introduction
Autism spectrum disorder is a complex developmental disorder that involves persistent challenges with social interaction, restricted interests, repetitive behaviors, impaired communication ability and deficits in social-emotional reciprocity1. As per Global Burden of Disease Study (GBD) autism affects 52.9 million children under the age of five2. According to WHO 2021 report, the majority of young children suffering from autism live in underdeveloped nations or low and middle-income countries. In South Asia and Sub-Saharan Africa, more than one million children suffer from ASD, and the Middle East, Central Asia, and North Africa have the highest rates of childhood autism3.
Children diagnosed with ASD at the age of two to four years are reported to have an accelerated total brain volume growth in the frontal and temporal region and as they reach the age of ten to fifteen years, the brain’s volumetric capacity declines4. ASD is referred to as a “spectrum” disorder since there is a wide diversification of its types and severity of symptoms5. One of the first indicators of ASD is a delay in language development and children with ASD commonly show impaired language development leading to a lack of social communication1. Autism is caused by a combination of inherited, nongenetic, and environmental factors, and the majority of them appear to influence key aspects of early brain development6. Some appear to affect the nerve cells interactions with one another in the brain, while others appear to influence how various areas of the brain communicate with one another7.
Variations in gene and genomes studies have identified an increasing number of SNPs associated with ASD over the past decade8,9. It has been estimated that SNPs explain between 17 and 60% of ASD, hence their contribution must not be overlooked10,11. The functions of the genetic variants responsible for the association with ASD are, however, poorly understood. A single nucleotide polymorphism (SNP) can lead to translation of the incorrect amino acid, resulting in defective or non-functional proteins. Such mutations in the lysine-specific methyltransferase 2H enzyme have been reported to alter histone methylation and affects brain development, increasing the risk of ASD and other neural disorders 12,13. Abnormal regulation of gene expression, disruption of proper neural development, and chromatin deregulation, results in aberrant chromatin remodeling. This interferes with the usual expression of genes essential in brain development, as well as abnormalities in synaptic adaptability and ultimately leads to ASD14.
In the present study, in silico analysis was carried out to identify different genes rare mutations associated with ASD as risk factors. These SNPs possess the potential to serve as important biomarkers for early detection of ASD, monitoring the course of ASD, and to develop possible drug therapies for ASD [Figure 1].
Methodology
Online bioinformatics tools were used to extract the data. Genetics Home Reference (GHR) database which is a MedlinePlus trusted health information, provided by National Library for Medicine was used to collect data of the genes whose mutations led to autism spectrum disorder which is found under the tab Genetics followed by Genetic Conditions15. Pathogenic and non-pathogenic mutant alleles associated with ASD were studied using the database retrieved from variation viewer tab under the related information column of National Center for Biotechnology Information (NCBI) site for the said gene which we found out from Genetics Home Reference database16. Location of the gene, families to which they belong, and function of the proteins encoded by them were analyzed by MedlinePlus database and documented. NCBI-gene site was used to identify transcript mutation and Rapid Stain Identification Series (rsIDs). The NCBI-Single Nucleotide Polymorphisms (SNP) database was used to examine and identify mutations in codon sequences that resulted in amino acid sequence changes. Variation and phenotype, clinical significance, and effect of the mutated genes on the amino acid sequence was recorded.
Result and Discussions
The current investigation discovered that ASD is associated with mutations in the activity dependent neuroprotector homeobox (ADNP), AT-rich interaction domain 1B (ARID1B), ASH1 like histone lysine methyltransferase (ASH1L), chromodomain helicase DNA binding protein 2 (CHD2), chromodomain helicase DNA binding protein 8 (CHD8), dual specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A), pogo transposable element derived with ZNF domain (POGZ), SH3 and multiple ankyrin repeat domains 3 (SHANK3), and synaptic Ras GTPase activating protein 1 (SYNGAP1) genes (Table 1).
Table 1: Table showing genes responsible for ASD, their location and function.
| GENES | LOCATION | NORMAL FUNCTION IN CELL |
| ADNP | Long arm (q) of Chromosome 20 band 13 (Location: 20q13.13) | Encodes for the protein involved in proper brain development by controlling the expression of other genes through chromatin remodeling. |
| ARID1B | Long arm (q) of Chromosome 6 band 25 (Location: 6q25.3)
|
Provides instructions to encode a protein composed of several SWI/SNF protein complexes, forming a subunit which regulates gene activity by chromatin remodeling and functions as a tumour suppressor. |
| ASH1L | Long arm (q) of Chromosome 1 band 22 (Location: 1q22) | Provides instructions for making a histone methyltransferase enzyme known as lysine-specific methyltransferase 2H. This enzyme stimulates the expression of genes involved in brain development. |
| CHD2 | Long arm (q) of Chromosome 15 band 26 (Location: 15q26.1) | This gene encodes a chromodomain DNA helicase protein 2 that regulates gene activity via chromatin remodeling. It aids in the modulation of neuron growth and function. |
| DYRK1A | Long arm (q) of Chromosome 21 (Location: 21q22.2) | DYRK1A is involved in phosphorylation and in the formation and maturation of dendritic spines from dendrites. |
| POGZ | Long arm (q) of chromosome 1 (Location: 1q21.3) | POGZ protein regulates gene expression by altering how tightly sections of DNA are packed by binding to chromatin for chromatin remodeling. |
| SHANK3 | Long arm (q) of Chromosome 22 (Location: 22q13.3) | SHANK3 protein ensures that the signals sent by one neuron are received by another. It serves as a framework for the connections between neurons and is also involved in dendritic spine development and maturation. |
| SYNGAP1 | Short arm (p) of chromosome 6 (Location: 6p21.32) | SynGAP controls neuronal adaptations, supports correct brain circuitry. |
Activity dependent neuroprotector homeobox (ADNP):
ADNP gene produces a neuro-protective peptide that operates during early embryogenesis, particularly during neurulation, and promotes glia-derived, survival-promoting chemicals that protect impaired nerve cells from cell death17, 18. ADNP gene is located on Chromosome 20 band 13’s long arm (q) and the encoded protein controls the expression of genes like BRG1 and CHD4, which are involved in normal brain development through chromatin remodeling19. The substantial neuroprotective activity of ADNP protein can be attributed entirely to the NAP domain, an octapeptide Asn-Ala-Pro-Val-Ser-Ile-Pro-Alaor NAPVSPIQ20. ADNP protein binds to DNA and interacts with SWI/SNF complexes by connecting the C-terminus to three of its key components throughout the remodeling process, which affect the shape of chromatin directly21,22. The majority of ADNP variants leads to the formation of short ADNP protein that can bind to DNA but not interact with SWI/SNF complexes. Variations at the terminus of a protein are typically unlikely to influence protein function23. Decreased ADNP expression in haploinsufficient populations has been shown to effectively deregulate the feedback loop by blocking wild-type protein from interacting to the promoter binding domain or occupying alternate target sequences23. These changes likely explain the intellectual disability in case of ASD. Around 24 mutations (small intragenic deletions, insertions, missense, and splice site variations) were found in ADNP Normal coding amino acids like tyrosine and leucine are mutated to terminating codons by frameshift mutations in ADNP gene, which may result in the creation of shortened proteins and disordered chromatin remodeling. Activation of many genes, as well as the development and function of several human tissues and organs, including the brain are all affected by chromatin remodeling disruptions. Furthermore, some of these mutations may prevent the production of neurotransmitters like epinephrine and norepinephrine, which are important for neural communication and mood regulation by encoding different amino acids. For example, a mutation of asparagine which is required for neurotransmitter production to lysine.
AT-rich interaction domain 1B (ARID1B)
ARID1B plays a pivotal role in controlling gene activity by forming a DNA-binding protein of the Brahma-associated factor chromatin remodeling complexes of numerous distinct SWI/SNF protein complexes which modulate gene activity by chromatin remodeling24, 25. ARID1B contains over 2,000 amino acid residues yet it has only two characterized protein domains, an AT-rich interaction domain (ARID) and domain of undefined function 3518 (DUF3518)22. ARID1B’s DNA-binding ability is impaired, and the BAF complex’s function is jeopardized as a result of missense mutations in the ARID domain26. In BAF complexes, the DUF3518 domain has been shown to interact with the BRG1 and BRM helicase subunits24. Because the ARID1B component may bind to DNA, it is thought to contribute in the targeting of SWI/SNF complexes to the chromatin ought to be remodeled24. Missense mutations in any of these domains can cause BAF complexes to become non – functional. Nonsense-mediated mRNA decay (NMD) is primarily triggered by nonsense and frameshift mutations, which cause premature translation termination27. As a result, these mutations result in the NMD of the ARID1B mRNA rather than the expression of mutant ARID1B protein. Truncating mutations that circumvent NMD typically result in a distinct and more extreme phenotype than the dominant negative impacts of the mutant protein25. Human learning and memory may be affected by frameshift mutations in the ARID1B gene, which replace glutamine with arginine, glycine, or serine. Similarly, mutations causing normal coding amino acids like threonine and alanine to a different branched amino proline is likely to disrupt the structure of the ARID1B protein. Stop-gain mutation of glycine affects immunomodulatory in the peripheral nervous system. ARID1B gene variants linked to ASD may lead to a reduction ARID1B protein level or a disturbance in the protein’s chromatin remodeling activity. As a result, the ARID1B gene plays an essential part in brain development. Further research needs to be conducted to determine whether individuals with ARID1B haploinsufficiency have altered brain development, which contributes to the intellectual disability and speech difficulty defined by ASD28,29.
Table 2: Table showing pathogenic alleles of the genes* responsible for ASD.
| GENE | rsID | AMINO ACID SEQ CHANGE | Consequence of mutation | VARIATION TYPE |
| ADNP | rs587777525 | Y [Tyr] ⇒ Ter[*] [AMB] | Frameshift variant | Indel |
| rs587777523 | N [Asn] ⇒ K [Lys] | Frameshift variant | Indel | |
| rs587777522 | L [Leu] ⇒ I [Ile] | Frameshift variant | Indel | |
| rs1057518978 | Y [Tyr] ⇒ V [Val] | Frameshift variant | Indel | |
| rs886042026 | K [Lys] ⇒ Q [Gln] | Frameshift variant | Indel | |
| rs779340209 | S [Ser] ⇒ Ter [*] [OPA] | Stop-gained | Single Nucleotide Variation | |
| rs1131691770 | L [Leu] ⇒ Ter [*] [AMB] | Stop Gained | Indel | |
| rs886041116 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs587777526 | Y [Tyr] ⇒ Y [Tyr] | Stop Gained | Single Nucleotide Variation | |
| rs1135401808 | Y [Tyr] ⇒ Ter [*] [OCH] | Stop gained | Indel | |
| rs886041449 | L [Leu] ⇒ P [Pro] | Frameshift variant | Deletion | |
| rs587777524 | S [Ser] ⇒ Ter [*] [AMB] | Stop gained | Single Nucleotide Variation | |
| ARID1B | rs587777114 | S [Ser] ⇒ P [Pro] | Intron Variant | Single Nucleotide Variation |
| rs867707366 | S [Ser] ⇒ L [Leu] | Stop gained | Single Nucleotide Variation | |
| rs387907143 | K [Lys] ⇒ Ter [*] [AMB] | Stop Gained | Single Nucleotide Variation | |
| rs750447037 | Q [Gln] ⇒ K [Lys] | Stop Gained | Single Nucleotide Variation | |
| rs797045282 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs886041819 | W [Trp] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs1028186690 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs797045283 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs1057517704 | Q [Gln] ⇒ E [Glu] | Stop Gained | Single Nucleotide Variation | |
| rs879253747 | Q [Gln] ⇒ Ter [*] [AMB] | Stop Gained | Single Nucleotide Variation | |
| rs902091666 | K [Lys] ⇒ E [Glu] | Stop Gained | Single Nucleotide Variation | |
| rs758570139 | Y [Tyr] ⇒ Y [Tyr] | Stop Gained | Single Nucleotide Variation | |
| rs387907144 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| ASH1L | rs863225241 | R [Arg] ⇒ Q [Gln] | Missense variant | Single Nucleotide Variation |
| rs1293246328 | A [Ala] ⇒ S [Ser] | Missense variant | Single Nucleotide Variation | |
| rs864309548 | Q [Gln] ⇒ Ter [*] [AMB] | Stop Gained | Single Nucleotide Variation | |
| rs398122998 | W [Trp] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs1057523601 | Q [Gln] ⇒ Ter [ *] [AMB] | Stop Gained | Single Nucleotide Variation | |
| rs797044912 | R [Arg] ⇒ R [Arg] | Stop Gained | Single Nucleotide Variation | |
| rs761127171 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs767106034 | R [Arg] ⇒ R [Arg] | Stop Gained | Single Nucleotide Variation | |
| rs777803897 | R [Arg] ⇒ R [Arg] | Stop Gained | Single Nucleotide Variation | |
| rs773860345 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs775406327 | S [Ser] ⇒ L [Leu] | Stop Gained | Single Nucleotide Variation | |
| rs2272457 | Y [Tyr] ⇒ Y [Tyr] | Stop Gained | Single Nucleotide Variation | |
| rs1057519228 | T [Thr] ⇒ T [Thr] | Synonymous Variant | Single Nucleotide Variation | |
| rs397514739 | T [Thr] ⇒ L [Leu] | Splice Donor Variant | Indel | |
| DYRK1A | rs886041658 | Y [Tyr] ⇒ Ter [*] [AMB] | Stop Gained | Single Nucleotide Variation |
| rs797044522 | D [Asp] ⇒ R [Arg] | Frameshift Variant | Indel | |
| rs886039652 | V [Val] ⇒ F [Phe] | Frameshift Variant | Indel | |
| rs797045042 | A [Ala] ⇒ G [Gly] | Frameshift Variant | Indel | |
| rs724159956 | R [Arg] ⇒ T [Thr] | Frameshift Variant | Indel | |
| rs1064794006 | K [Lys] ⇒ N [Asn] | Frameshift Variant | Indel | |
| rs797045539 | P [Pro] ⇒ T [Thr] | Frameshift Variant | Insertion | |
| rs724159953 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs797044520 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs797045041 | R [Arg] ⇒ Q [Gln] | Missense variant | Single Nucleotide Variation | |
| rs1064796713 | N [Asn] ⇒ I [Ile] | Frameshift Variant | Indel | |
| POGZ | rs1235715459 | P [Pro] ⇒ S [Ser] | Frameshift Variant | Indel |
| rs869312834 | Q [Gln] ⇒ Ter [*] [AMB] | Stop Gained | Single Nucleotide Variation | |
| rs869320764 | E [Glu] ⇒ T [Thr] | Frameshift Variant | Deletion | |
| rs864321667 | L [Leu] ⇒ C [Cys] | Frameshift Variant | Indel | |
| rs796052217 | Q [Gln] ⇒ R [Arg] | Frameshift Variant | Deletion | |
| rs1085307702 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs864321673 | S [Ser] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs1057518170 | M [Met] ⇒ V [Val] | Frameshift Variant | Deletion | |
| rs869320763 | E [Glu] ⇒ Ter [*] [OPA] | Frameshift Variant | Indel | |
| rs142133690 | Y [Tyr] ⇒ Ter [*] [AMB] | Stop Gained | Single Nucleotide Variation | |
| rs864321675 | L [Leu] ⇒ F [Phe] | Frameshift variant | INDEL | |
| SHANK3 | rs886041467 | P [Pro] ⇒ L [Leu] | Frameshift Variant | Indel |
| rs1294272918 | Q [Gln] ⇒ E [Glu] | Stop Gained | Single Nucleotide Variation | |
| rs886041430 | L [Leu] ⇒ R [Arg] | Frameshift Variant | Indel | |
| rs762292772 | A [Ala] ⇒ P [Pro] | Frameshift Variant | Indel | |
| rs761720914 | E [Glu] ⇒ K [Lys] | Stop Gained | Single Nucleotide Variation | |
| rs886041238 | R [Arg] ⇒ L [Leu] | Frameshift Variant | Indel | |
| rs1238131472 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs1057519395 | S [Ser] ⇒ R [Arg] | Frameshift Variant | Indel | |
| rs1064793514 | L [Leu] ⇒ R [Arg] | Frameshift Variant | Deletion | |
| rs1131691727 | P [Pro] ⇒ H [His] | Frameshift Variant | Indel | |
| rs1064795759 | Q [Gln] ⇒ Ter [*] [AMB] | Stop Gained | Single Nucleotide Variation | |
| rs121918316 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| SYNGAP1 | rs587780470 | L [Leu] ⇒ C [Cys] | Frameshift Variant | Indel |
| rs1131691635 | L [Leu] ⇒ S [Ser] | Frameshift Variant | Indel | |
| rs1060503386 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs1057519400 | S [Ser] ⇒ Q [Gln] | Frameshift Variant | Indel | |
| rs1060503383 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| Rs797046029 | L [Leu] ⇒ P [Pro] | Frameshift Variant | Indel | |
| rs797046028 | V [Val] ⇒ Ter [*] [AMB] | Stop gained | Indel | |
| rs1057519405 | P [Pro] ⇒ R [Arg] | Frameshift Variant | Indel | |
| rs397514670 | P [Pro] ⇒ L [Leu] | Missense variant | Single Nucleotide Variation | |
| rs1131692154 | R [Arg] ⇒ Ter [*] [OPA] | Stop Gained | Single Nucleotide Variation | |
| rs121918315 | K [Lys] ⇒ Ter [*] [OCH] | Stop Gained | Single Nucleotide Variation |
*(only some data is provided)
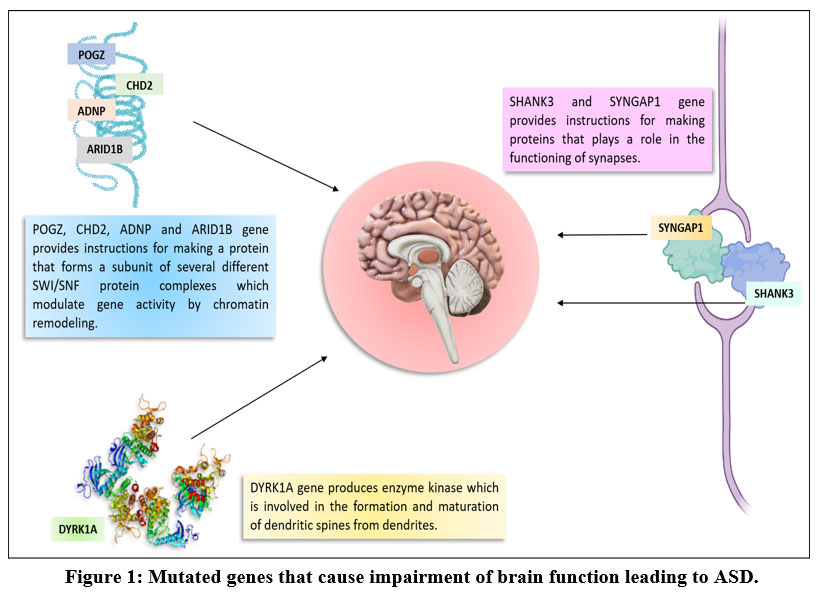 |
Figure 1: Mutated genes that cause impairment of brain function leading to ASD. |
ASH1 like histone lysine methyltransferase (ASH1L)
The lysine-specific methyltransferase 2H enzyme is produced byASH1L gene and can be found in a range of organs and tissues throughout the body31. Lysine-specific methyltransferase 2H functions as a histone methyltransferase enzyme that modify histone proteins. Histone methyltransferases control the function of few genes by methylation of histone27. Additionally, certain genes involved in brain development are activated by lysine-specific methyltransferase 2H. Histone methylation is disrupted by the lack of a functioning lysine-specific methyltransferase 2H enzyme32. Loss of ASH1L gene has been shown to impair embryonic and postnatal brain development, as well as the formation of neuronal networks in the developing hypothalamus, which is required for optimal feeding behaviors and initial postnatal growth32, 33. At least seven ASH1L gene mutations were found to be associated with ASD. Some ASH1L gene mutations linked to ASD induce a single amino acid alteration in the lysine-specific methyltransferase 2H enzyme, while others disrupt genetic material from the ASH1L gene sequence or produce an early stop signal, rendering the enzyme inactive and affecting neural connection. For example; when alanine to serine and arginine to glutamine missense mutations occur in ASH1L gene, alanine production is impaired, which is a source of energy in the CNS, and the body’s metabolism is also affected as arginine production is hindered.
Chromodomain helicase DNA binding protein 2 (CHD2)
CHD2 gene codes for chromodomain DNA helicase protein 2 (CHD2) which controls gene activity through chromatin remodeling and is present in all cells of the body34. This protein plays a significant role in the function and growth of neurons present in the brain. It is proposed that the protein translated from the mutant CHD2 gene causes enhanced neuronal excitability due to the action of GABAergic neuron excitability or other electrophysiological channels and cause convulsions, and have little or no effect on RNA expression levels35. CHD2 related neurodevelopmental disorders are autosomal dominant disorders caused by a de novo pathogenic variant. Twenty-seven CHD2 gene variants linked to ASD were investigated. These mutations may result in moderate alterations in the expression of multiple genes, due to their induced functional disruption of the CHD2 protein, all of which affect development of brain and increase the possibility of ASD. For example, valine is used for energy production while serine helps in biosynthesis of purines and pyrimidines. The frameshift mutation in CHD2 gene of valine and serine results in a stop codon that completely disrupts chromatin remodeling and can possibly lead to ASD.
Dual specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A)
DYRK1A encodes a kinase enzyme involved in the phosphorylation of proteins which helps in regulating the activity of proliferation and differentiation of cells and play important role in the development of the nervous system31. Dendritic spines assist convey nerve impulses and facilitate communication between neurons, and the DYRK1A enzyme is involved in their production and maturation from dendrites in neurons36. A nonsense mutation results in the synthesis of a kinase domain protein with a C-terminally shortened C-terminus (DYRK1A-E396ter), which is then processed by the proteasome into an inactive form, demonstrating overall loss-of-function of DYRK1A37. Anxiety, microcephaly (an abnormally small head), intellectual disabilities, and speech issues are other specific traits seen in ASD patients with DYRK1A gene mutations38. Chronic seizures (epilepsy), distinctive facial features, poor muscular tone (hypotonia), foot deformities, and walking impairments may be experienced by these individuals38. At least twenty-five DYRK1Agene mutations associated with ASD were identified. DYRK1A enzyme dysfunction or absence causes aberrant gene expression regulation and affects normal brain development. For example, frameshift mutation in DYRK1A gene changes normal coding tyrosine to a termination codon which can lead to production of a truncated protein which in turn can have many determinantal effects on the brain.
Pogo transposable element derived with ZNF domain (POGZ)
POGZ proteins are zinc finger proteins which are able to bind to the chromatin for chromatin remodeling owing to its structure which consist of a unique pattern of amino acids and one or more zinc ions and they are capable of binding. Zinc finger domain’s folded form stabilizes the protein, and allows it to interact with many other molecules39. POGZ protein modulates gene expression, which is critical for brain development, by interacting with the SP1 transcription factor, heterochromatin protein 1 (HP1), and chromodomain helicase DNA-binding protein 4 (CHD4) and acting as a chromatin regulator 40, 41. The large majority of previously reported de novo POGZ gene mutations in patients with neurodevelopmental disorders (NDDs) are nonsense and frameshift mutations dispersed between the C2H2 Zn finger and centromere protein-B-like DNA-binding (CENP-DB) domains, as well as within the CENP-DB domain itself. The large majority of reported de novo POGZ genetic variants in patients with neurodevelopmental disorders (NDDs) are nonsense and frameshift mutations dispersed between the C2H2 Zn finger and centromere protein-B-like DNA-binding (CENP-DB) domains, including within the CENP-DB domain itself 42. Twenty-two pathogenic mutations in the POGZ gene were investigated, which may result in a POGZ protein with reduced chromatin binding ability, causing aberrant chromatin remodeling and eventually compromising the normal expression of genes essential in brain development42. Mutations in UTR-5′ and frameshift mutations in the POGZ gene can affect expression level and mRNA translation kinetics by producing DNA binding elements or cis-regulatory motifs.
SH3 and multiple ankyrin repeat domains 3 (SHANK3)
SHANK3 protein is vital in the working of synapses because it works as a platform that maintains the connections between neurons, ensuring that signals sent by one neuron are recognized by another43. The N-terminal region of SHANK3 comprising of the Shank/ProSAP N-terminal (SPN) domain and a set of ankyrin (Ank) repeats has been reported with several missense mutations44. These mutations impact the Abi1 binding site in SHANK3, which is the core location of the SHANK protein, and missense mutations cause alterations outside of established interacting motifs in the C-terminal region of SHANK proteins, making it impossible to evaluate the precise role of these mutations44. In the N-terminal motif of Shank/ProSAP (SPN), which is essential in dendritic spine growth and maturation, seven ankyrin repeats (Ank) have been identified as a hotspot for missense mutations45. It has already been reported that disruption in communication between neurons contributes to the development of ASD46. A minimum of 24 SHANK3gene mutations associated with ASD were identified out of which most of them impairing SHANK3 protein function or synthesis. For example, mutation of proline to alanine impedes the assembly of several proteins and the rate of peptide bond production by the ribosome, cds-synon mutation of arginine and tyrosine, frameshift mutation of glutamic acid into a stop codon that handicaps memory and focus power, and missense mutation of isoleucine into phenylalanine may directly affects gene function.
Synaptic Ras GTPase activating protein 1 (SYNGAP1)
SynGAP protein is encoded by SYNGAP1 gene, located in synapses in the brain, and regulates important biochemical signaling pathways that enable learning and memory47. SynGAP regulates synapse adaptations and promotes proper brain architecture by linking nerve cells, and it is especially vital during a crucial stage of early developing brain that influences long-term psychological capability48. Pre-mRNA splicing in the SYNGAP1 gene is dependent on the concordance of “cis” sequences that define exon-intron boundaries, which is required for proper protein translation, and regulatory sequences recognized by splicing machinery. Improper exon and intron recognition reported by point mutations at these consensus sequences may lead to the development of an abnormal transcript of the affected gene45. At least thirty SYNGAP1gene mutations were identified, these mutations hampered or completely blocked the function or production of SynGAP protein associated with ASD. These mutations may be the underlying cause of behavioral impairments associated with ASD. For example, frameshift mutation of amino acids (tryptophan, leucine, valine and arginine) to stop codons and also mutations affecting normal coding the branched-chain amino acids like leucine, isoleucine, and valine with multiple functions in the brain can affect the synaptic plasticity in the brain.
Conclusion
The relevance of SNPs in ASD-associated genes as a risk factor was investigated in this study using in silico analysis. Mutations in the ADNP, ARID1B, ASH1L, CHD2, and POGZ genes have been identified to disrupt the development and functioning of the brain by altering gene regulation and causing abnormal chromatin modeling. Many of the SNP mutations studied result in a premature stop signal, resulting in a nonfunctional enzyme that is exceptionally short. Furthermore, these mutations in other genes associated with ASD, SHANK3 and SYNGAP1 disrupt the function of synapses and, as a result, cell-to-cell communication. These mutations may cause changes in synaptic adaption, which may exacerbate the behavioral problems associated with ASD. These SNPs can act as biomarkers and provide insight into the etiology of ASD. Furthermore, the SNPs identified above may aid in the development of ASD medical interventions. More examination and validation of the identified SNPs will be required to assess their clinical importance and applicability in translation research as novel targets.
Acknowledgment
This work was supported by DBT STAR College grant and Gargi College, University of Delhi.
Conflict of Interest
The authors have no conflict of interest to declare.
Funding Sources
Not Applicable
References
- Hodges, H., Fealko, C., & Soares, N. (2020). Autism spectrum disorder: definition, epidemiology, causes, and clinical evaluation. Pediatr., 2020; 9(Suppl 1), S55-S65. https://www.mhlw.go.jp/content/10500000/000704736.pd.
CrossRef - World Health Organization (WHO), Autism Spectrum Disorder https://www.who.int/news- room/fact-sheets/detail/autism-spectrum-disorders.
- Ha, S., Sohn, I. J., Kim, N., Sim, H. J., & Cheon, K. A. Characteristics of Brains in Autism Spectrum Disorder: Structure, Function and Connectivity across the Lifespan. Neurobiol., 2015; 24(4), 273–284. https://doi.org/10.5607/en.2015.24.4.273.
CrossRef - National Institute of Health, U.S. Department of National Institute of Neurological Disorders and Stroke https://www.nimh.nih.gov/ health/topics/autism-spectrum-disorders-asd.
- National Research Council (US) and Institute of Medicine (US) Committee on Integrating the Science of Early Childhood Development, Shonkoff, J. P., & Phillips, D. A. (Eds.). From Neurons to Neighborhoods: The Science of Early Childhood Development, 2000. National Academies Press (US).
- Ratajczak H. V. Theoretical aspects of autism: causes–a review. J. Immunotoxicol. 2011; 8(1), 68–79. https://doi.org/10.3109/ 1547691X.2010.545086.
CrossRef - Ha, S., Sohn, I. J., Kim, N., Sim, H. J., & Cheon, K. A. Characteristics of Brains in Autism Spectrum Disorder: Structure, Function and Connectivity across the Lifespan. Exp. Neurobiol., 2015; 24(4), 273–284. https://doi.org/10.5607/en.2015.24.4.273.
CrossRef - Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431444.
CrossRef - Robinson EB, iPSYCH-SSI-Broad Autism Group, St Pourcain B, Anttila V, Kosmicki JA, Bulik-Sullivan B, et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nature Genetics. 2016;48:552–555.
CrossRef - Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism. 2012;3:9.
CrossRef - Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–885.
CrossRef - Pandey, P.; Sharma, P. Analysis of Early Onset of Alzheimer’s Disease Genes: Disease Causing and Risk Factors. Eur. J. Biol. Res., 2021;11, 251-259. http://dx.doi.org/10.5281/zenodo.4641962
- Koemans TS, Kleefstra T, Chubak MC, et al. Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet., 2017;13(10):e1006864. https://doi.org/10.1371/journal.pgen.1006864
CrossRef - Hoffmann, A., & Spengler, D. Chromatin Remodeler CHD8 in Autism and Brain Development. J. Clin. Med., 2021; 10(2), 366. https://doi.org/10.3390/jcm10020366
CrossRef - https://medlineplus.gov/genetics/condition/autism-spectrum-disorder/
- https://www.ncbi.nlm.nih.gov/variation/view/
- Bassan, M., Zamostiano, R., Davidson, A., Pinhasov, A., Giladi, E., Perl, O., Bassan, H., Blat, C., Gibney, G., Glazner, G., Brenneman, D. E., & Gozes, I. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J. Neurochem., 1999; 72(3), 1283–1293. https://doi.org/10.1046/j.1471-4159.1999.0721283.x
CrossRef - Said S. I. Molecules that protect: the defense of neurons and other cells. Clin. Investig., 1996; 97(10), 2163–2164. https://doi.org/10.1172/JCI118655.
CrossRef - Arnett, A. B., Rhoads, C. L., Hoekzema, K., Turner, T. N., Gerdts, J., Wallace, A. S., Bedrosian-Sermone, S., Eichler, E. E., & Bernier, R. A. The autism spectrum phenotype in ADNP syndrome. Autism Res., 2018; 11(9), 1300–1310. https://doi.org/10.1002/aur.1980
CrossRef - Magen, I., & Gozes, I. Davunetide: Peptide therapeutic in neurological disorders. Med. Chem., 2014; 21(23), 2591–2598. https://doi.org/10.2174/0929867321666140217124945
CrossRef - Vandeweyer G, Helsmoortel C, Van Dijck A, et al. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am J Med Genet C Semin Med Genet., 2014;166C(3):315-326. https://doi.org/10.1002/ajmg.c.31413
CrossRef - Helsmoortel C, Vulto-van Silfhout AT, Coe BP, et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet., 2014;46(4):380-384. doi:10.1038/ng.2899
CrossRef - Kapitansky O, Gozes I. ADNP differentially interact with genes/proteins in correlation with aging: a novel marker for muscle aging. Geroscience., 2019;41(3):321-340. https://doi.org/10.1007/s11357-019-00079-x
CrossRef - Sim JC, White SM, Lockhart PJ. ARID1B-mediated disorders: Mutations and possible mechanisms. Intractable Rare Dis Res., 2015;4(1):17-23. https://doi.org/10.5582/irdr.2014.01021
CrossRef - Wang X, Nagl NG, Wilsker D, et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J., 2004;383:319-325. https://doi.org/10.1042/BJ20040524
CrossRef - Inoue H, Furukawa T, Giannakopoulos S, Zhou S, King DS, Tanese N. Largest subunits of the human SWI/SNF chromatin-remodeling complex promote transcriptional activation by steroid hormone receptors. J Biol Chem., 2002;277(44):41674-41685. https://doi.org/10.1074/jbc.M205961200
CrossRef - Hurlstone AF, Olave IA, Barker N, van Noort M, Clevers H. Cloning and characterization of hELD/OSA1, a novel BRG1 interacting protein. Biochem J., 2002;364(Pt 1):255-264. https://doi.org/10.1042/bj3640255
CrossRef - Schweingruber C, Rufener SC, Zünd D, Yamashita A, Mühlemann O. Nonsense-mediated mRNA decay – mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta., 2013;1829(6-7):612-623.. https://doi.org/10.1016/j.bbagrm.2013.02.005
CrossRef - Halgren C, Kjaergaard S, Bak M, et al. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin Genet., 2012;82(3):248-255. https://doi:10.1111/j.1399-0004.2011.01755.x
CrossRef - ASH1L gene, Genetic Home Reference.
- Gao Y, Duque-Wilckens N, Aljazi MB, et al. Loss of histone methyltransferase ASH1L in the developing mouse brain causes autistic-like behaviors. Commun Biol., 2021;4(1):756. https://doi.org/10.1038/s42003-021-02282-z
CrossRef - Cheon S, Culver AM, Bagnell AM, et al. Counteracting epigenetic mechanisms regulate the structural development of neuronal circuitry in human neurons. Mol Psychiatry., 2022;10.1038/s41380-022-01474-1. https://doi.org/10.1038/s41380-022-01474-1
CrossRef - Nieto-Estevez V, Hsieh J. CHD2: One Gene, Many Roles. Neuron. 2018;100(5):1014-1016. https://doi.org/10.1016/j.neuron.2018.11.036
CrossRef - Carvill GL, Mefford HC. CHD2-Related Neurodevelopmental Disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; December 10, 2015.
- Nourbakhsh K, Yadav S. Kinase Signaling in Dendritic Development and Disease. Front Cell Neurosci., 2021;15:624648. https://doi.org/10.3389/fncel.2021.624648
CrossRef - Martinez de Lagran M, Benavides-Piccione R, Ballesteros-Yañez I, et al. Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cereb Cortex., 2012;22(12):2867-2877. https://doi.org/10.1093/ cercor/bhr362
CrossRef - Lee KS, Choi M, Kwon DW, et al. A novel de novo heterozygous DYRK1A mutation causes complete loss of DYRK1A function and developmental delay. Sci Rep., 2020;10(1):9849. https://doi.org/10.1038/s41598-020-66750-y
CrossRef - Levy JA, LaFlamme CW, Tsaprailis G, Crynen G, Page DT. Dyrk1a Mutations Cause Undergrowth of Cortical Pyramidal Neurons via Dysregulated Growth Factor Signaling. Biol Psychiatry., 2021;90(5):295-306. https://doi.org/10.1016/j.biopsych.2021.01.012
CrossRef - Markenscoff-Papadimitriou E, Binyameen F, Whalen S, et al. Autism risk gene POGZ promotes chromatin accessibility and expression of clustered synaptic genes. Cell Rep., 2021;37(10):110089. https://doi.org/10.1016/j.celrep.2021.110089.
CrossRef - Tan B, Zou Y, Zhang Y, et al. A novel de novo POGZ mutation in a patient with intellectual disability. J Hum Genet., 2016;61(4):357-359. https://doi.org/10.1038/jhg.2015.156
CrossRef - Suliman-Lavie, R., Title, B., Cohen, Y., Hamada, N., Tal, M., Tal, N., Monderer-Rothkoff, G., Suliman-Lavie R, Title B, Cohen Y, et al. Author Correction: Pogz deficiency leads to transcription dysregulation and impaired cerebellar activity underlying autism-like behavior in mice. Nat Commun., 2021;12(1):3874. https://doi.org/10.1038/s41467-021-24166-w
CrossRef - Suliman-Lavie R, Title B, Cohen Y, et al. Author Correction: Pogz deficiency leads to transcription dysregulation and impaired cerebellar activity underlying autism-like behavior in mice. Nat Commun., 2021;12(1):3874. https://doi.org/10.1038/s41467-020-14697-z
CrossRef - Halbedl S, Schoen M, Feiler MS, Boeckers TM, Schmeisser MJ. Shank3 is localized in axons and presynaptic specializations of developing hippocampal neurons and involved in the modulation of NMDA receptor levels at axon terminals. J Neurochem., 2016;137(1):26-32. https://doi.org/10.1111/jnc.13523.
CrossRef - Woike D, Wang E, Tibbe D, et al. Mutations affecting the N-terminal domains of SHANK3 point to different pathomechanisms in neurodevelopmental disorders. Sci Rep., 2022;12(1):902. https://doi.org/10.1038/s41598-021-04723-5.
CrossRef - Hassani Nia F, Kreienkamp HJ. Functional Relevance of Missense Mutations Affecting the N-Terminal Part of Shank3 Found in Autistic Patients. Front Mol Neurosci., 2018;11:268. https://doi.org/10.3389/fnmol.2018.00268.
CrossRef - Vyas Y, Cheyne JE, Lee K, Jung Y, Cheung PY, Montgomery JM. Shankopathies in the Developing Brain in Autism Spectrum Disorders. Front Neurosci., 2021;15:775431. https://doi.org/10.3389/fnins.2021.775431.
CrossRef - Gamache TR, Araki Y, Huganir RL. Twenty Years of SynGAP Research: From Synapses to Cognition. J Neurosci., 2020;40(8):1596-1605. https://doi.org/10.1523/JNEUROSCI.0420-19.2020.
CrossRef - Brimble E, Lee-Messer C, Nagy PL, Propst J, Ruzhnikov MRZ. Clinical Transcriptome Sequencing Confirms Activation of a Cryptic Splice Site in Suspected SYNGAP1-Related Disorder. Mol Syndromol., 2019;9(6):295-299. https://doi.org/10.1159/000492706
CrossRef

