Elucidation of Computational 3D Models of Protein Drug Targets for Colletotrichum Falcatum a Fungal Plant Pathogen Causing Red Rod of Sugarcane
 , Sabia Imran and Lokesh Ravi*
, Sabia Imran and Lokesh Ravi* Department of Botany, St.Joseph’s College (Autonomous), Bengaluru-27
Corresponding Author E-mail : Lokesh.ravi@sjc.ac.in
DOI : http://dx.doi.org/10.13005/bpj/1926
Download this article as:
![]()
Objective of this study is to develop 3D structures of the potential drug target proteins in the ascomycte plant pathogen Colletotrichum falcatum that causes ‘red rot disease’ a grave disease of sugarcane crop. This study uses online databases such as UniPort, DrugBank, PDB and PMDB to retrieve and submit biological information. Online webtools such as SwissModel, BLASTp, was used to construct homology models and find similarity, respectively. Total of 72 protein sequences were identified as potential drug targets and were retrieved form UniProt in .fasta file format. Based on the available template model, total of 52 proteins were successfully modelled using Swiss-Model webtool. Among these 52 predicted models, 41 models were identified as significant based on Ramachandran plot analysis. The 41 predicted models were submitted to PMDB server for public access. This study has created a dataset of 3D homology models of drug target proteins that can greatly benefit in selective drug discovery and drug development against the investigated pathogen Colletotrichum falcatum. This can greatly benefit the pharmaceutical industry in developing agricultural antifungal favouring sugarcane cultivation.
KEYWORDS:Colletotrichum Falcatum; Homology Modeling; PMDB; Ramachandran Plot; Red Rot of Sugarcane; Swiss-Model
Introduction
The Red-Rot Disease
Saccharum officinarium (sugarcane) is a monocotyledonous plant species that belongs to the family Graminae and is cultivated in most of the tropical and subtropical regions of the world [1]. It is the 2nd most valuable agro-industrial crop (cash-crop) in India next to cotton [2, 3]. It occupies around 4.2%(4.36million hectares) of the total area under cultivation and contributes 7.5% to the gross value of agricultural production of the country [1, 2]. Global contributors to sugarcane production include Brazil, India, China, Thailand, the US, and the UK. India is the 2nd largest producer of sugarcane in the world following Brazil and is the largest consumer of sugarcane with an average consumption of 19 million MT a year[3].Sugarcane is economically very important as it stores a high concentration of sucrose in its stalk tissues, but several biotic and abiotic factors affects this sucrose yield. Approximately a hundred diseases have been reported globally, with 100 fungi, 10 bacteria,10 viruses, and 50 nematode species known to cause destructive diseases to sugarcane [3]. The most devastating disease of sugarcane is the red rot disease, caused by the fungi Colletotrichum falcatum.
Colletotrichum Falcatum
It is a facultative saprophyte which belongs to the subdivision Ascomycotina [3]. It causes huge loss of 18-31% in sugarcane production. According to the GOI study of 2017, various biotic stress factors, including red rot disease hampers the production of sugarcane in many important parts of the country [3][4]. The pathogen affects the economically valuable stalk by entering through the nodes [5]. Meteorological factors, along with soil pH and waterlogging play a major, but a compound role in aiding the infection mechanism [3, 6–9]. Susceptibility of the cultivar, along with the age of stalk tissues and time of infestation, determines the symptoms shown by the plant [7]. Infections decrease juice purity and deplete important micro and macronutrients like iron, copper, zinc, potassium, and phosphorous, but however, increase the content of calcium, and nitrogen to a large extent [10, 11]. Stalk weight is reduced by 29% furthermore, sugar retrieval capacity is reduced by up to 31%. Affected tissues cannot be commercially utilized because they lead to deterioration of the product [3, 11].
Homology Modeling
In this study, the 3D structures of proteins of Colletotrichum falcatum, are developed using homology modeling technique. These structures are necessary to design and develop drugs that could aim to stop the spread of this dreadful disease.Lack of protein structures has hindered the understanding of binding specificities of proteins and ligands, which are pre-requisites for drug design and development[12]. Well established and recognized databases and tools were employed in this study to obtain the computational structures of potential drug target proteins. These are very necessary to contain this dreadful disease and also reduce the monetary burdens incurred on the farmers due to this disease. Homology modelling was done using the pre-existing fasta sequences of the proteins and structure of the protein was predicted based on the available templates which were similar to the respective query sequence of the protein [13][14][15]. It is of utmost importance that the protein structures are available, which would otherwise hinder the understanding of binding specificities of the protein and ligand leading to low availability of drugs to stop the spread of this disease. The structures developed in this study could further be exploited to design and develop new and efficient drugs which would be a boon to the cultivators of this crop.
Materials and Methods
NCBI Database
This database was used to screen the amount of pre-existing information available on the chosen organism of interest. A simple search with the organism’s name revealed the amount of available data in different databases (https://www.ncbi.nlm.nih.gov/).
Sequence Retrieval
The UniProtknowledgebase (www.uniprot.org)-a centralised, reliable and publically accessable collection of non–reductant protein sequences [16], was used to retrieve the amino acid sequences required for this study. The organism’s name was used as the search criteria. The best sequences were sorted based on the length of the amino acid chain and were downloaded in.fasta file format. Repetitive sequences were all omitted to prevent ambiguity, and downloaded sequences were all stored along with their respective accession IDs.
Sequence Alignment
The query sequences or amino acid sequences retrieved from UNIPROT were all compared using the BLASTp server (www.uniprot.org), an online tool that compares the query sequence with pre-existing protein sequences in the Protein Data Bank in order to obtain the percentage similarity [17][14][18]. The first BLAST was carried out to get similarity values with pre-existing sequences of the non-reductant database, followed by a second BLAST to get similarity values with the sequences present in the Protein Data Bank (www.rcsb.org). The similarity estimations thus obtained in percentages were noted down for further reference.
Structure Prediction
The 3D structure of the selected drug target proteins were predicted through homology modelling technique, using the online tool SWISS-MODEL (https://swissmodel.expasy.org/). This tool uses respective amino acid sequences of the protein as well as the templates available in the protein databank to predict the 3D structure of the protein[19][20].Sequences retrieved from UNIPROT were uploaded in .fasta file format and the predicted 3D models were retrieved in .pdb file format. The quality of the models developed was dependent on the availability and percentage similarity of the templates.
The models hence developed were analyzed using the Ramachandran plots. These are graphical plots that represent protein structures in terms of torsion angles andhence play a significant role in confirming the predicted structure’s accuracy [21]. The best models were downloaded in .pdb after the analysis was complete.
Model Analysis
Qualitative analysis of the reliability of the predicted model was performed via the Ramachandran plot provided by the SWISS-MODEL as an in-built feature. The degree angles of all the residues were expected to be within the Most-Favoured regions of the Ramachandran plot, that determines the quality of the predicted structure. The residues that are outside of this favoured region are considered to be unfavourable prediction or outliers. These outliers reduce the confidence score of the predicted model.
Model Submission
The 3D models thus predicted with good quality Ramachandran plot (confidence score) were then submitted to public database Protein Model Data Base [PMDB] (https://bioinformatics.cineca.it/PMDB/) which is a resource that stores manually built protein models that are published in scientific literature [22]. All the models were uploaded in .pdb file formats and each entry was given a unique PMDBID for future reference.
Results and Discussions
Drug Target Selection
A bibliographical search of the organism Colletotrichum falcatum in NCBI website, showed that, there are no known protein structures of the organism, but there are about 600 protein sequences reported. Hence, this organism was ideal for construction of computational protein models. Preliminary screening of the 600 protein sequences, suggested that non-enzymatic protein components such as ribosomal subunits were predominantly reported. Hence, these ribosomal and non-enzymatic proteins were eliminated for the homology model development, resulting in a total of 125 protein sequences for further analysis. These 125 protein sequences were then analysed in the DrugBank database (www.drugbank.ca) to confirm if they are previously reported as a possible drug targets. Among these 125 protein sequences, 72 sequences were identified as potential drug targets based on literature proof of mechanism of action of known antibiotics.
Building Homology Model
The selected 72 protein sequences were retrieved from the uniprot database (www.uniprot.org) and saved as fasta file format (.fasta) then subjected for BLASTp also known as Protein Blast (https://blast.ncbi.nlm.nih.gov/) to search within Protein Data Bank (www.rcsb.org) to identify the templates for homology modelling. Protein structures with more than 80% sequence similarity were chosen as ideal template. Only 12 sequences had a similarity of more than 80%. The other sequences had more than 1 template used for homology modelling, with less than 80% sequence similarity. All the 72 protein sequences with appropriate template models were subjected for homology model construction using the Swiss-Model web server tool (https://swissmodel.expasy.org/). Homology protein models were built for 52 sequences successfully.
Ramachandran Plot Validation
The Swiss-Model tool, has developed a maximum of 5 different models for each of the query sequences. The best of the 5 models were selected based on the Ramachandran Plot analysis, that was integrated with the server. The ramachandran plot of all developed models were examined, and the model which has majority percentage of the residues within the most preferred regions of the graph were considered as the ideal / final model. The percentage of residues within the most favoured regions was also used as the confidence percentage score. The graphical representation of the ramachandran plot of least preferred model and best preferred models with lowest and highest confidence score are shown in Figure.1(A) & Figure.1(B). The structure analysis of a constructed homology model for WRKY transcription factor 37 protein is represented graphically in Figure.2.
![Figure 1: Ramachandran plot analysis of; [A]: Least preferred model with lowest confidence score of 79.3% (Coronative insensitive protein homolog-1); [B]: Most preferred model with highest confidence score of 100% (WRKY transcription factor 37)](https://biomedpharmajournal.org/wp-content/uploads/2020/04/Vol13No2_Elu_Ani_Fig1-150x150.jpg) |
Figure 1: Ramachandran plot analysis of; |
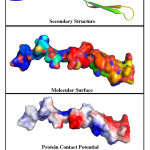 |
Figure 2: Structural analysis of the most preferred homology model with 100% |
Submission to PMDB
A total of 52 homology models were developed in the Swiss-Model online tool. However, ramachandran plot analysis showed that only 41 protein models exhibited more than 90% residues within the most favoured regions. Hence, these 41 sequences were submitted in the Protein Model Data Base (https://bioinformatics.cineca.it/PMDB/) for public access and further research. A summary of all the 41 protein 3D models submitted to the PMDB database with their PMDB ID, along with drug target name, Uniprot ID and confidence score of the model are tabulated in Table.1.
Table 1: Summary of PMDB entries for Colletotrichum falcatum
| Sl. No | Drug Target Name | UniProt ID | Confident Score | PMDB ID |
| 1 | Chitin synthase | J7HAD5 | 91.89 | PM0082471 |
| 2 | Beta-tubulin(142) | Q8J1X0 | 95.16 | PM0082472 |
| 3 | Elongation factor 2 | A0A1B3B2L9 | 91.43 | PM0082473 |
| 4 | Dirigent protein | A0A1B3B2N1 | 96.97 | PM0082474 |
| 5 | EPL1 protein(212) | A0A1C9II66 | 98.25 | PM0082310 |
| 6 | NAC transcription factor C | A0A059VE62 | 95.65 | PM0082475 |
| 7 | NAC transcription factor J | A0A059VK69 | 91.49 | PM0082476 |
| 8 | NAC transcription factor M | A0A059VPC0 | 92.68 | PM0082477 |
| 9 | Glycosyl hydrolase family 10 | A0A1C9II18 | 98.68 | PM0082478 |
| 10 | WRKY transcription factor 37 | A0A059VP87 | 100 | PM0082479 |
| 11 | Elongation factor 1 alpha | A0A1B3B2J0 | 97.22 | PM0082480 |
| 12 | Translation elongation factor 1 alpha | A0A1B3B2K3 | 94.74 | PM0082481 |
| 13 | BZIP transcription factor 25 | A0A059VP32 | 99.64 | PM0082482 |
| 14 | BZIP transcription factor 9 | A0A059VJB5 | 100 | PM0082483 |
| 15 | BZIP transcription factor 15 | A0A059VP28 | 100 | PM0082484 |
| 16 | BZIP transcription factor 18 | A0A059VK55 | 100 | PM0082491 |
| 17 | BZIP transcription factor 29 | A0A059VE58 | 95.65 | PM0082492 |
| 18 | BZIP transcription factor 4 | A0A059VE50 | 98 | PM0082493 |
| 19 | BZIP transcription factor 24 | A0A059VP99 | 96.23 | PM0082494 |
| 20 | BZIP transcription factor 14 | A0A059VP93 | 92.31 | PM0082495 |
| 21 | TLP transcription factor A | A0A059VJE2 | 91.11 | PM0082497 |
| 22 | TLP transcription factor K | A0A059VK79 | 98.96 | PM0082498 |
| 23 | TLP transcription factor M | A0A059VJE8 | 93.62 | PM0082499 |
| 24 | TLP transcription factor L | A0A059VE75 | 93.62 | PM0082500 |
| 25 | MYB transcription factor 83 | A0A059VP36 | 93.27 | PM0082501 |
| 26 | MYB transcription factor 82 | A0A059VPA7 | 96.2 | PM0082502 |
| 27 | MYB transcription factor 78 | A0A059VJC5 | 90.54 | PM0082503 |
| 28 | Class VII chitinase | A0A1B3B2M2 | 98.68 | PM0082504 |
| 29 | Chitinase B | A0A1B3B2K5 | 95.89 | PM0082505 |
| 30 | RNA -dependent RNA polymerase | A0A3Q9NNK8 | 92.64 | PM0082309 |
| 31 | Putative heat shock protein (90) | A0A1B3B2M4 | 91.43 | PM0082506 |
| 32 | Respiratory burst oxidase-like protein H | A0A1B3B2N7 | 98.44 | PM0082507 |
| 33 | NDR1/HIN1-like protein | A0A1B3B2P4 | 100 | PM0082508 |
| 34 | Type I polyketide synthase | C9WLC4 | 93.83 | PM0082509 |
| 35 | Respiratory burst oxidase-like protein B | A0A1B3B2N2 | 95.45 | PM0082510 |
| 36 | Putative ethylene response sensor | A0A1B3B2N0 | 100 | PM0082511 |
| 37 | Beta-1,3-glucanase D | A0A1B3B2K9 | 97.06 | PM0082512 |
| 38 | EPL1-like protein | A0A1C9II08 | 96 | PM0082513 |
| 39 | Salicylate hydroxylase 1 | A0A1C9IIB1 | 95.45 | PM0082514 |
| 40 | Bys1 family protein | A0A1C9II96 | 97.14 | PM0082515 |
| 41 | Respiratory burst oxidase-like protein C | A0A1B3B2N8 | 97.25 | PM0082516 |
Conclusion
The objective of this study was to prepare 3-D models of proteins present in Colletotrichum falcatum that is a major pathogen responsible for the red rot disease in sugarcane crops. The protein structures of this ascomycete were successfully developed and analyzed using the swiss-model workspace. A similar study by Divya et.al (2018) was performed on a less studied organism P.marinus an endoparasitic pathogen, and other Perkinsus spp. that are responsible for causing devastating losses in the cultivation of shellfish and mollusk species worldwide , where the authors developed and submitted 3D structures of drug target proteins of the organism based on homology modelling [23] .
These developed structures could be further exploited to either develop new antifungal drugs or to revise the pre-existing ones. This is an important aspect of computer-aided, in-silico drug designing approach which gained significant momentum in the recent years [24]. Furthermore active sites of the molecule can be found out aiding in docking studies that would help in studying the interactions between the protein and the ligand. A Study by Daisy et.al (2013) involving the in-silico drug designing approach for biotin protein ligase of Mycobacterium tuberculosis, provides better insight on the significant role played by predicted protein structures in the field of drug design and development [25].
The structures can also prove to be useful for understanding the mechanism of infection of the disease. They can also be used to design a pathway for understanding the effect of inhibition of a few proteins that can act as good antifungal drug targets which may help design an antifungal compound that might be more effective as well as non-toxic unlike the traditional fungicides and insecticides that are proven to be harmful in the long run. This study provides a basic platform for future in-silico work on Colletotrichum falcatum specific drug design and development.
Acknowledgement
The authors thank the management of St. Joseph’s College (Autonomous), Bengaluru for supporting this research work.
Conflict of Interest
None declared.
References
- Gawade DB, Pawar BH, Gawande SJ, Vasekar VC, Antagonistic Effect of Trichoderma Against Fusarium moniliformae the Causal of Sugarcane Wilt Plant Pathology Section , Division of Crop Protection ,. 12(9): 1236–1241 (2012)
- Jayashree J, Selvi A, Nair N V, Characterization of Resistance Gene Analog Polymorphisms in sugarcane cultivars with varying levels of red rot resistance. 1(July): 1191–1199 (2010)
- Sharma R, Tamta S, A Review on Red Rot : The ” Cancer ” of Sugarcane. Plant Pathol Microbiol, (2015)
- Varma PK, Kumar KVK, Suresh M, Kumar NR, Sekhar VC, Potentiality of Native Pseudomonas spp . in Promoting Sugarcane Seedling Growth and Red Rot ( Colletotrichum falcatum went ) Management. 7(02): 2855–2863 (2018)
- Viswanathan R, Ramasamy S, Viswanathan , R . and R . Samiyappan ( 2000 ). Red rot disease in sugarcane : Challenges and prospects . Madras Agricultural. (January): 549–559 (2000)
- Kumar A, Sharma SK, Management of red rot of sugarcane caused by colletotrichum falcatum went : A review. 7(3): 5029–5039 (2019)
- Patel P, Krishnamurthy R, Physiological profiling of Colletotrichum falcatum , the causal agent of Sugarcane Red rot disease Physiological profiling of Colletotrichum falcatum , the causal agent of Sugarcane Red. (October) (2017)
- Paswan S, Kumar M, Sattar A, Impact of Weather Factors on Development of Red Rot Disease of Sugarcane Agro-Ecosystem. 7(02): 8–12 (2018)
- Phytopathology I, Agnihotri VP, Current sugarcane disease scenario and management strategies.
- Gupta RN, Sah SB, Kumar S, Kumar A, Kishore C, Chand G, Impact of Red Rot Disease on Nutrient Status of Sugarcane. 44(7): 3533–3538 (2018)
- Kamat DN, Losses Due to Red Rot Pathogen in Cane Juice Quality. 7(02): 13–16 (2018)
- Name C, Name S, Cycle L, Pacific Pests and Pathogens – Fact Sheets Sugarcane red rot ( 221 ). (221)
- Cavasotto CN, Phatak SS, Homology modeling in drug discovery : current trends and applications. 14(July) (2009)
- Article R, Homology Modeling a Fast Tool for Drug Discovery : Current Perspectives. 1–17
- Scientific R, International Journal Of. 7(3) (2016)
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, et al., UniProt : the Universal Protein knowledgebase. 32 (2004)
- Mcginnis S, Madden TL, BLAST : at the core of a powerful and diverse set of sequence analysis tools. 32: 20–25 (2004)
- Manuscript A, NIH Public Access. 7(3): 217–227 (2007)
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al., SWISS-MODEL : modelling protein tertiary and quaternary structure using evolutionary information. 42(April): 252–258 (2014)
- Arnold K, Bordoli L, Schwede T, Structural bioinformatics The SWISS-MODEL workspace : a web-based environment for protein structure homology modelling. 22(2): 195–201 (2006)
- Kumar P, Arya A, Ramachandran Plot : A simplified approach. (December) (2018)
- Meo PDO De, Cozzetto D, The PMDB Protein Model Database. (January) (2006)
- Jindam D, Ravi L, Krishnan K, Construction of computational protein database by homology modeling for the aquatic pathogen Perkinsus marinus.
- Wadood A, Ahmed N, Shah L, Ahmad A, Hassan H, Shams S, In-silico drug design: An approach which revolutionarised the drug discovery process. OA Drug Des Deliv, 1(1): 3 (2013)
- Daisy P, Nivedha RP, Bakiya RH, In silico drug designing approach for biotin protein ligase of mycobacterium tuberculosis. Asian J Pharm Clin Res, 6(SUPPL.1): 103–107 (2013)




