Translating Molecular Pathogenesis into Therapeutics: Emerging Strategies and Clinical Prospects for Alzheimer’s Disease
 , Shivani Thakur 1, Pankaj Kumar 2, Sourbh Suren Garg 3, Ankush Goyal 1and Pradeep Goyal4
, Shivani Thakur 1, Pankaj Kumar 2, Sourbh Suren Garg 3, Ankush Goyal 1and Pradeep Goyal4 1School of Pharmacy, Maharaja Agrasen University, Baddi, India
2Department of Pharmacology, University Institute of Pharmaceutical Sciences, Panjab University, Chandigarh, India
3Advance Centre of Research and Innovation, CGC University, Mohali, India
4Department of Pharmacology, Saraswati College of Pharmacy Gharuan, Mohali, India
Corresponding Author Email: piplanimona44@gmail.com
Download this article as:
ABSTRACT:Alzheimer’s disease (AD) is a devastating neurodegenerative disorder that affects millions of people worldwide and poses a major global health challenge. This complex disease involves multiple pathogenic factors, including amyloid-beta (Aβ), tau protein aggregation, synaptic dysfunction, mitochondrial impairment, oxidative stress, and genetic influences. While the intricate interplay and individual contributions of these elements to AD progression remain under investigation, considerable advancements have been made in elucidating their roles. This review examines the molecular mechanisms underlying the pathogenesis of AD, highlighting how each factor contributes to the onset and progression of the disorder. To date, significant efforts in AD research have focused on diverse therapeutic approaches, including targeting Aβ and tau proteins, modulating neuroinflammation, enhancing autophagy, and restoring synaptic function. As these strategies evolve, they provide renewed hope for interventions that could slow AD progression and improve cognitive function. Furthermore, a multi-targeted approach addressing these interconnected pathways shows promise for managing the complex pathology of AD. Continued research in these areas will be essential for identifying novel therapeutic options and advancing clinical strategies for AD management.
KEYWORDS:Alzheimer’s disease; Amyloid beta; Mitochondrial impairment; Neuroinflammation; Oxidative stress
Introduction
Alzheimer’s disease (AD) is an escalating global health crisis, expected to affect over 107 million individuals by 2050.1 This neurodegenerative disorder, primarily afflicting older adults, devastates not only the cognitive and physical abilities of those affected but also impacts their emotional well-being, financial resources, and social support networks.2 As global populations age at an unprecedented rate, dementia in older adults, particularly AD, has emerged as a significant health challenge that demands urgent attention. With its complex pathology involving amyloid-beta (Aβ) deposits, tau protein aggregation, synaptic dysfunction, mitochondrial impairment, oxidative stress, and genetic factors, AD presents numerous therapeutic challenges. This disease is named after Alois Alzheimer, the neurologist who first discovered it, and his work laid the foundation for subsequent research into AD’s pathogenesis, beginning with the identification of Aβ plaques as a key contributing factor.3 Subsequently, Iqbal et al. (1974) isolated neurofibrillary tangles (NFTs) from the brains of patients with AD.4 As research has advanced, numerous hypotheses have emerged to explain the complex mechanisms of AD. Extensive studies have shown that AD pathogenesis is linked to Aβ deposition, tau protein phosphorylation, cholinergic dysfunction, neuroinflammation, and mitochondrial oxidative stress.5-8 Despite ongoing research, currently available treatments for AD offer only limited symptom relief, highlighting the urgent need for innovative therapeutic approaches.9 The prevalence of dementia caused by AD presents a significant health and social challenge across both developed and developing nations.10
In AD, aggregates of Aβ proteins form senile plaques, while NFTs arise from the accumulation of tau proteins.1 The disease is influenced by various factors, including advancing age, genetic predisposition, vascular diseases, head injuries, and infections. Current treatment options are primarily limited to cholinesterase inhibitors (such as Donepezil and Galantamine) and N-methyl D-aspartate (NMDA) receptor antagonists like Memantine and Amantadine. It is important to note that these drugs do not provide a cure or prevent disease progression.11
While several genes are known to contribute to the early onset of Alzheimer’s, the fundamental causes of the more common late-onset form remain uncertain. Unfortunately, effective treatments are limited for the 35 million Americans currently affected by Alzheimer’s, underscoring the need for continued research to better understand AD’s underlying processes and to discover novel treatment approaches that could pave the way for innovative therapies in the future.2 This comprehensive review delves into the intricate molecular mechanisms underlying AD, focusing on the complex interactions between Aβ and tau proteins, mitochondrial dysfunction, neuroinflammation, and synaptic impairment. While numerous reviews have addressed various aspects of AD, this article offers a critical analysis of previously published studies, providing an in-depth evaluation of emerging drug candidates that are still in the early stages of clinical trials, yet hold promise for clinical approval and widespread use. By exploring these molecular pathways and therapeutic approaches, this review not only sheds light on the pathogenesis of AD but also highlights the potential for novel treatment strategies aimed at halting or reversing disease progression. These insights could pave the way for more effective interventions for the growing population affected by this debilitating neurodegenerative disorder.
Molecular mechanisms of AD
Hyperphosphorylation of tau proteins and Aβ proteins is recognised as a factor for AD, but the severity of this disorder is more directly linked to cognitive impairments and disruptions in neurotransmission. The precise role of these characteristics in the disease’s progression is a topic of debate. Nevertheless, extensive research in biochemistry, cell biology, molecular biology, and pathology has provided valuable insights into how the aggregates of hyperphosphorylated tau proteins and Aβ proteins may have a direct effect on synapses, which may finally alter neurotransmission.12
In AD, Aβ plays a central role and has a wide-ranging impact on cellular processes within the brain, contributing significantly to neurodegeneration. Aβ is involved from its initial generation through amyloid precursor protein (APP) processing to its interactions within cells. It disrupts organelle function, triggers harmful cellular responses, and leads to tau protein hyperphosphorylation, forming neurofibrillary tangles. Aβ also disrupts mitochondrial function, calcium regulation, protein homeostasis, autophagy, transcription regulation, and inflammation, all of which complicate AD pathophysiology and contribute to disease progression.13 The protective function of the blood-brain barrier (BBB) is to safeguard the brain, ensuring that its environment remains secure. However, in cases of neuroinflammation, the BBB may be compromised, posing potential risks to the central nervous system. While animal studies provide valuable insights, human models are essential for accurately reflecting human pathophysiology.14
AD is closely associated with synaptic dysfunction, a major contributor to cognitive decline. Aβ disrupts synaptic integrity by impairing glutamate receptors, altering calcium homeostasis, and triggering glial activation. Activated microglia and astrocytes release pro-inflammatory cytokines that dysregulate glutamate, causing excitotoxicity. Aβ also impairs mitochondrial function and critical signalling pathways, leading to energy deficits that worsen synaptic damage. Moreover, Aβ synergizes with tau pathology, amplifying neurotoxicity and cognitive decline, underscoring the complex interplay between these hallmark features of AD.15 Since mitochondria are essential for supporting synaptic function and play a pivotal role in the context of this neurodegenerative ailment, continued research has increasingly emphasized the intricate connection between Aβ, tau, and mitochondrial dysfunction. However, many studies have shown that the impairment of mitochondria is closely associated with synaptic failure and the neurodegeneration observed in AD.16
Oxidative stress and impaired mitochondrial function represent pivotal factors in the pathogenesis of numerous neurodegenerative disorders, including Parkinson’s disease, depression, Huntington’s disease, and epilepsy. The effects of oxidative stress result from an overproduction of reactive oxygen species, which harm mitochondria in multiple ways, including causing DNA mutations, disrupting respiratory chain function, and disturbing calcium regulation. These mitochondrial issues significantly contribute to neurodegenerative diseases by exacerbating neuronal dysfunction and initiating neurodegeneration.17 In the context of AD, genetically acquired forms have been associated with three causative genes: Aβ precursor protein, presenilin 1, and presenilin 2 (APP, PSEN1, and PSEN2), particularly in relation to the ε4 allele of the apolipoprotein E (APOE) gene. The discovery of these genes has paved the way for the development of numerous animal studies, which have played an essential role in advancing our understanding of the causes of AD.18 The various mechanisms involved in AD are summarized in Figure 1 and Table 1.
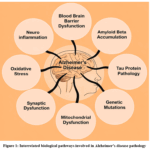 |
Figure 1: Interrelated biological pathways involved in Alzheimer’s disease pathology |
Table 1: Major molecular mechanisms implicated in AD and their contributions to disease pathology
| Mechanism/pathway | Contribution to disease symptoms |
| Aβ accumulation | Excess Aβ plaques generated from APP via β- and γ-secretase form toxic oligomers and fibrils, disrupting neurons and triggering neuroinflammation. 12, 13 |
| Tau protein pathology | Hyperphosphorylated tau forms NFTs, impairing axonal transport and synaptic function, leading to neuronal death. 13 |
| Neuroinflammation | Aggregated Aβ activates microglia to release pro-inflammatory cytokines, exacerbating neuronal damage. 13, 15 |
| Oxidative stress | ROS overproduction damages lipids, proteins, and DNA, contributing to neurodegeneration. 17 |
| Synaptic dysfunction | Aβ oligomers impair synaptic plasticity and neurotransmission, causing cognitive deficits. 15 |
| Genetic mutations | Mutations in APP, PSEN1, and PSEN2 genes are associated with familial AD. 18 |
| BBB dysfunction | BBB disruption allows peripheral immune cells and proteins to enter, promoting neuroinflammation. 14 |
| Mitochondrial dysfunction | Impaired mitochondria lead to synaptic failure and neuronal death. 16 |
Amyloidogenic pathway: accumulation of Aβ and proteolytic cleavage of APP (amyloid precursor protein)
The accumulation of Aβ is an indicator of AD pathology and plays a crucial role in the neurodegenerative cascade. This process begins with the proteolytic cleavage of the APP, leading to the formation of Aβ peptides. The breakdown of the APP is facilitated by enzymes known as secretases. Certain secretases, such as β-secretase and γ-secretase, generate the harmful Aβ-peptide, while another enzyme, α-secretase, interrupts its production by cleaving the protein in the middle of the harmful segment.19 Imbalance between the production and clearance of Aβ42 plays a crucial early role in the development of AD.20 These secretase enzymes operate within cells, and the β-APP traverses the cell’s transport system to reach the sites where these enzymes are active. The β-secretase, primarily active in the trans-golgi network and endosomes, initiates the amyloidogenic pathway by cleaving APP at the N-terminus.21 This cleavage produces a soluble β-carboxyl-terminal fragment and a C-terminal fragment (β-CTF) known as C99 or β-CTF. Subsequently, γ-secretase, a multi-subunit protein complex anchored in cellular membranes, cleaves β-CTF within the transmembrane domain, generating Aβ-peptides of various lengths, with Aβ42 being particularly prone to aggregation and considered more neurotoxic. Additionally, brain activity and certain gene mutations may also influence the production of the Aβ-peptide.22 Dysregulated processing leads to the accumulation of Aβ-peptides, which aggregate into toxic oligomers and fibrils. These Aβ aggregates impair synaptic function, disrupt neuronal signalling, and initiate a cascade of pathological events, ultimately contributing to neuroinflammation, synaptic dysfunction, and neuronal death.23 In the case of γ-secretase, the identification of presenilin as the catalytic site has been pivotal, with dominant mutations causing early-onset AD occurring in either the substrate APP or the protease involved in Aβ generation. Additionally, apolipoprotein E4 has been linked to impaired Aβ clearance from the brain.24
Aβ generation from γ-secretase-mediated cleavage of the neuronal transmembrane APP is a central event in AD. The reasons behind the accumulation of Aβ in the brains of elderly individuals remain unclear, but they may involve changes in how APP is metabolized or how Aβ is cleared.25 Aβ exists in two forms, Aβ40 and Aβ42, with Aβ42 being more prone to aggregation. There may also be a protective mechanism: α-secretase, an enzyme that cleaves APP in the Aβ region, prevents the formation of Aβ. However, the impact of cell density on APP processing and Aβ production is less understood. It has been observed that decreased cellular density significantly increases levels of Aβ40, Aβ42, total Aβ, and the Aβ42:β40 ratio, highlights the crucial role of cell density in APP processing. These findings provide strong evidence that there could be significant implications for studies on conditions, genetics, and drugs affecting APP function and Aβ production.26
Aggregated Aβ peptides and neurotoxic effects
Elevated levels of Aβ peptides in the brain are associated with a decline in cognitive function. In the case of sporadic forms of the disease, the primary issue appears to be insufficient clearance of Aβ proteins, whereas familial AD is linked to increased production of these proteins. A crucial event in AD is the aggregation of Aβ, leading to its deposition. However, the precise factors that contribute to driving Aβ aggregation and accumulation in sporadic AD are not fully understood.27 Aggregated Aβ peptides induce neurotoxicity by activating microglia and astrocytes, triggering chronic neuroinflammation that worsens neuronal damage. Misfolded proteins are recognized by glial receptors, stimulating innate immune responses and the release of inflammatory mediators that drive disease severity. Genetic factors affecting plaque clearance, along with conditions like obesity and systemic inflammation, further impair immune function and accelerate AD progression.28
Aβ neurotoxicity involves multiple molecular factors, including membrane lipids, receptors, channels, signalling pathways, cytoskeletal proteins, and inflammatory mediators. Soluble Aβ oligomers, alongside insoluble plaques, interact with various membrane components, disrupting synaptic function, neuronal communication, and network integrity, while also inducing inflammation and oxidative stress.29 Figure 2 illustrates the amyloidogenic pathway. Multidisciplinary research has established this pathway as a central feature of AD pathophysiology, enabling the identification of therapeutic targets, particularly in early disease stages. Nevertheless, further study of its physiological roles and upstream regulators is needed to inform comprehensive prevention strategies.30
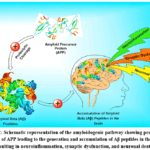 |
Figure 2: Schematic representation of the amyloidogenic pathway showing proteolytic cleavage of APP leading to the generation and accumulation of Aβ peptides in the brain, resulting in neuroinflammation, synaptic dysfunction, and neuronal death. |
Tau protein pathology
In vitro studies indicate that senile plaques have direct cytotoxic effects. This influences neurotransmission, signalling pathways, the functions of various organelles, and immunity, ultimately leading to synaptic loss and impairment in neurotransmitter release. Evidence from studies such as Rajmohan and Reddy (2017) suggests that, despite the complex pathophysiology of these lesions, their removal could mitigate disease severity and progression.12 Tau protein pathology plays a pivotal role in the pathogenesis of AD. Tau is a microtubule-associated protein primarily found in neurons and becomes a central player in this condition due to abnormal post-translational modifications, mainly phosphorylation. This aberrant phosphorylation results in the formation of neurofibrillary tangles, which are a hallmark pathological feature of AD.31 Tau proteins are insoluble filaments that accumulate as NFTs in AD and function by maintaining the structural stability of microtubules. However, in a diseased brain, tau undergoes abnormal hyperphosphorylation. This leads to the disassembly of microtubules, and the released tau molecules aggregate to form paired helical filaments. Research indicates that the hyperphosphorylation of tau is likely due to disruptions in cell signalling, generally resulting from imbalances in the activities of various enzymes, such as phosphatases and kinases. In AD, it appears that Aβ peptide plays a significant role in triggering these imbalances.32
The consequences of NFTs formation are profound. These NFTs impede axonal transport, a critical process for transporting vital molecules to various parts of the neuron. Disrupted axonal transport leads to the degeneration of axons, synaptic dysfunction, and a decline in neuronal communication. As a result, affected neurons lose their ability to maintain synaptic connections and ultimately succumb to cell death. Tau pathology is closely linked to cognitive decline in AD, with increased levels of NFT deposition correlating with the severity of dementia.33 Further research suggests that tau proteins may spread throughout the brain in a prion-like manner, which may exacerbate disease progression.34
Tau pathology is strongly associated with cognitive impairment in AD. A study using positron emission tomography (PET) imaging revealed that tau accumulation in specific brain regions correlates with cognitive decline, largely independent of amyloid deposits and only partially influenced by grey matter atrophy. These results suggest that tau-related cognitive deficits arise through multiple mechanisms, informing future clinical trials targeting tau pathology.35 In AD, the complex process of pathogenesis begins several years prior to the onset of any significant symptoms. The hallmark characteristics of the disorder, such as senile plaques and neurofibrillary tangles, gradually propagate throughout the brain. It has been shown that tau and Aβ proteins form NFTs and senile plaques through misfolding and self-assembly due to templated conformational changes. This process increases the production of toxic substances.36
Current data also suggest that the diffusion of these pathological lesions from one brain region to another is facilitated by the internalization, transport, and release of naturally occurring seeds composed of the same proteins. Abnormal phosphorylation of tau protein, which leads to the formation of NFTs, is a crucial mechanism in the pathogenesis of AD. These NFTs disrupt axonal transport, synaptic function, and neuronal integrity, ultimately contributing to cognitive decline and neurodegeneration.37 Different stages of the tau protein pathway have been illustrated in Figure 3.
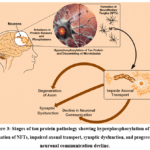 |
Figure 3: Stages of tau protein pathology showing hyperphosphorylation of tau, formation of NFTs, impaired axonal transport, synaptic dysfunction, and progressive neuronal communication decline. |
Neuroinflammation
Neuroinflammation in AD involves complex interactions between misfolded protein aggregates and the brain’s immune system. The binding of these proteins to glial receptors activates innate immunity and inflammatory responses. Genetic factors affecting protein clearance, along with external influences like obesity and chronic inflammation, further impair immune function and accelerate disease progression.28 Microglia-driven inflammation plays a key role in AD. Around Aβ deposits, microglia become activated, releasing chemokines and neurotoxic cytokines that damage the CNS, while also clearing Aβ and providing neuroprotection. Recent studies reveal that microglial functions are complex, involving diverse activation states and responses to pathogens.38
AD is marked by neuronal death and memory impairment, driven by apoptosis, autophagy, and necroptosis. Senile plaques and aggregated tau amplify neuroinflammation, with necroptosis and NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome activity further promoting the release of inflammatory mediators. This uncontrolled cell death disrupts neuronal communication and exacerbates disease progression.39 Neuroinflammation also arises in response to various damage signals, including injuries, infections, oxidative stress, the presence of redox-active iron, and accumulated toxic proteins.40 The mechanisms contributing to neuroinflammation are illustrated in Figure 4.
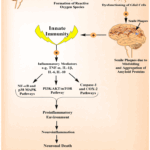 |
Figure 4: Neuroinflammation cascade in AD demonstrating how impaired brain homeostasis and ROS formation lead to glial dysfunction, amyloid plaque formation, activation of innate immunity, release of inflammatory mediators, and progression toward neuronal death. |
In AD, activated microglia release various cytokines, leading to the production of a pro-inflammatory environment in the brain, which can directly damage neurons and impair their function.41 A study by Rani and colleagues attempted to investigate the role of pro-inflammatory cytokine levels in AD. They examined scopolamine-induced amnesia using an ELISA-based method to measure the levels of TNF-α and interleukins in the blood serum and various tissues (renal, cerebral, and hepatic). Their findings revealed that the cerebral tissue of AD-affected mice exhibited elevated levels of IL-1β, IL-6, IL-10, and TNF-α, suggesting the significant involvement of these pro-inflammatory cytokines in the development of AD.42 Meanwhile, another group of leading scientists indicated that several cytokines, including IL-6 and TNF-α, increase the activity of BACE1 and the expression of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) in the brains of patients, resulting in the production of Aβ, a crucial contributor to this disease. The aggregated proteins are attracted to the inflammatory site via chemotaxis. Various molecular pathways that trigger microglia to release inflammatory cytokines include caspase, nitric oxide, COX, NFκB, p38 MAPK, and IP3/Akt/mTOR.43 Similar to these findings, in vivo studies suggest that systemic inflammation occurring outside the CNS may play an important role in promoting neurodegeneration, the pathology of AD, and cognitive decline in older individuals. Pro-inflammatory cytokines released into the body can cross the BBB and promote a pro-inflammatory environment in the CNS. These cytokines may further stimulate inflammatory proteins through endothelial signalling and activation of the vagus nerve. Systemic infections can also induce reactive, inflammatory microglia and astrocytes, promote the oligomerization of Aβ, activate the complement system, and lead to the conversion of neurotransmitters into harmful bioactive metabolites following breakdown. These molecular modifications may be responsible for the initiation or exacerbation of neurodegenerative processes that can lead to dementia in vulnerable older patients.44
Neuroinflammation is a key contributor to AD severity, even at early stages, driven by abnormally activated microglia. Peripheral immune cells, such as macrophages and T cells, can infiltrate the brain, disrupting communication between adaptive and innate immune responses and exacerbating disease progression.45 Recent research based on transcriptomic data collected from the hippocampus using the Gene Expression Omnibus database revealed a significant role of immune cell infiltration in altering key genes responsible for the development of AD. Four key genes linked to AD were KDELR1, SPTAN1, CDC16, and RBBP6.46 Immune cells, particularly T cells, exhibit dysfunction and play a crucial role in AD pathology. Recent studies have demonstrated that abnormal T cells indirectly contribute to neuroinflammation through the release of pro-inflammatory compounds that directly interact with infiltrating glial cells in the brain. Various factors such as α-secretase, apolipoprotein E, tau protein, and β-secretase may influence the activation of T cells.47 Moreover, recent advancements in genetic research techniques have provided valuable insights into the relationship between inflammation and AD. Genetic variations related to inflammation have been linked to an increased risk of AD. These genetic variations are associated with various substances, including TNF-α, IL-6, IL-10, IL-4, IL-1, and transforming growth factor beta. The investigation of these genetic variants and their impact on inflammation may further elucidate the mechanisms underlying AD development.48,49
Oxidative stress and mitochondria dysfunction
AD is characterized by TNF-α-driven neuroinflammation, Aβ and tau pathology, and mitochondrial dysfunction, which together promote oxidative stress, synaptic failure, and neurodegeneration. Excess reactive oxygen species (ROS) further damage mitochondria by causing DNA mutations, disrupting the respiratory chain, altering membrane permeability, and impairing calcium homeostasis, mechanisms also implicated in other neurodegenerative disorders such as ALS.17
Oxidative stress contributes to Aβ accumulation and NFT formation by hyperphosphorylated tau. It arises from an imbalance between oxidants and antioxidants, leading to excessive ROS such as superoxide, hydrogen peroxide, and hydroxyl radicals. These ROS damage lipids, proteins, and nucleic acids, with the brain being especially vulnerable due to its high lipid content and oxygen consumption (Figure 5).50 AD involves significant oxidative stress even before overt pathology, with early mitochondrial structural and functional damage. Impaired mitochondria produce more ROS but less ATP, creating a vicious cycle in which mitochondrial dysfunction and oxidative stress mutually reinforce each other, amplifying neuronal damage.51
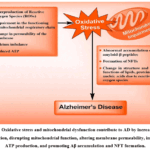 |
Figure 5. Oxidative stress and mitochondrial dysfunction contribute to AD by increasing ROS production, disrupting mitochondrial function, altering membrane permeability, impairing ATP production, and promoting Aβ accumulation and NFT formation. |
Mitochondrial dysfunction is central to AD progression, affecting energy supply, oxidative stress, and neuronal protection. Key altered mechanisms include mitochondrial biogenesis, dynamics, axonal transport, ER-mitochondria interactions, mitophagy, and proteostasis. Studying these processes offers promising avenues for understanding AD pathogenesis and developing potential therapies.52 Neurodegenerative disorders often involve cell bioenergetics dysfunction, with AD being the most common. Mitochondria play a central role in disrupted energy utilization. A study by Troutwine et al., 2022, demonstrates that mitochondrial dysfunction in Alzheimer’s disease is closely associated with classical pathological features, particularly Aβ accumulation, and occurs within the broader context of established risk factors such as tau pathology and APOE genotype.53
Mitochondrial dysfunction, manifested by ROS overproduction, calcium imbalance, reduced ATP, and impaired dynamics plays a central role in AD pathogenesis. Interventions such as exercise, antioxidant-rich diets, and targeted therapies that restore mitochondrial function may delay onset and slow disease progression. Addressing mitochondrial health and oxidative stress is thus essential, as highlighted by Misrani et al. (2021), in shaping current and future AD therapeutic strategies.54
AD is complex, and maintaining healthy mitochondria is crucial. While Aβ and tau proteins receive significant attention, restoring mitochondrial health is a promising strategy. Understanding the mechanisms involved in mitochondrial impairment is vital for exploring various aspects of mitochondrial regulation, offering opportunities for therapeutic interventions.55 Investigating mitochondrial dysfunction presents several challenges. The “mitochondrial cascade hypothesis” suggests that accumulating DNA mutations with age impact brain function. Declines in key regulatory mechanisms may initiate a cascade of deficits, leading to mitochondrial impairment and disease.56 Preclinical and clinical efforts have concentrated on the amyloid pathway, but clinical trials have not shown clear benefits.57 Few therapies specifically target mitochondrial impairment, focusing instead on reversing oxidative stress and neuronal death pathways. Recent research studies examining various bioenergetics and mitochondrial characteristics aim to provide suitable treatment approaches. Improving cellular bioenergetics in preclinical models may yield widespread beneficial effects for individuals suffering from AD.58
Current treatment for AD
Considering the multifaceted nature of AD and the complexity of its underlying mechanisms, developing effective therapeutic strategies remains a significant challenge. Therapeutic approaches to treat AD can be broadly classified into three categories: symptomatic, disease-modifying, and regenerative.59 Currently, only four Food and Drug Administration (FDA)-approved drugs from the symptomatic class are available for the treatment of AD. Three of these are inhibitors of the acetylcholinesterase enzyme (AChE), donepezil, galantamine, and rivastigmine, while one is an N-methyl-D-aspartate (NMDA) receptor antagonist, memantine.60 These treatment options are symptom-based rather than curative, aimed at limiting the progression of cognitive symptoms and behavioral and psychological symptoms of dementia, as well as other symptoms such as agitation, aggression, hallucinations, delusions, and insomnia.59,61 Additionally, both of these drug groups are approved as treatment options for patients in the moderate stage of AD 62, as these medications primarily address cognitive impairment and dysfunction in global activities in symptomatic AD.62,63 These treatments are delivered through the oral or transdermal route.61 The current treatment options for AD are discussed in detail below:
AChE inhibitors
According to the cholinergic hypothesis, in AD, the progressive loss of cholinergic innervation in the limbic and neocortical regions of the brain is responsible for the decline in memory, learning, attention, and other higher cognitive functions. Neurofibrillary degeneration in the basal forebrain is likely the primary cause of dysfunction and death of cholinergic neurons in this area, leading to widespread presynaptic cholinergic denervation.64 AChE inhibitors work by binding to nicotinic (nAChRs) and muscarinic (mAChRs) acetylcholine receptors, helping to sustain acetylcholine levels by preventing its hydrolysis. This regulation of neurotransmission increases synaptic acetylcholine levels, potentially enhancing the cognitive capacity of AD patients.62
Although there are differences in the pharmacokinetics and pharmacodynamics of the three approved drugs in this class, they do not exhibit significant differences in efficacy.60 Donepezil and galantamine function as selective and reversible inhibitors of acetylcholinesterase, while rivastigmine serves as a “pseudo-irreversible” inhibitor of both acetylcholinesterase and butyrylcholinesterase. Donepezil has a long elimination half-life of 70 h, whereas galantamine’s half-life ranges from 6 to 8 h.63,64 In contrast, rivastigmine has a shorter elimination half-life (1-2 h for oral administration and 3-4 h for transdermal administration), but it provides a prolonged duration of action, inhibiting acetylcholinesterase and butyrylcholinesterase for approximately 8.5 and 3.5 h, respectively.64 Since 2007, rivastigmine has been available in a transdermal patch form, which has been beneficial in reducing caregiver burden and improving treatment adherence.59 Overall, these inhibitors are well tolerated by patients with AD.60
Early initiation of AChE inhibitors after AD diagnosis is recommended, as delayed treatment accelerates cognitive decline. Common side effects include diarrhoea, nausea, vomiting, and, occasionally, REM sleep behaviour disorder. Taking medication after breakfast can reduce adverse effects, while rivastigmine patches may cause a local rash. These drugs can also induce bradycardia and syncope, making them unsuitable for patients with severe cardiac issues, active peptic ulcers, gastrointestinal bleeding, or uncontrolled seizures. Gradual dose escalation over months or years is advised to reach the maximum tolerated dose.64
NMDA receptor antagonist
Glutamate, a key neurotransmitter, is essential for learning and memory functions in the brain.61 However, elevated levels of glutamate due to overactivity can lead to excessive calcium (Ca2⁺) influx, which is implicated in neuronal death and various pathological effects.59,61 To manage these elevated glutamate levels, anti-glutamatergic drugs function as non-competitive antagonists at NMDA receptors. These treatments are designed to delay disease progression, stabilize or temporarily improve cognitive functions, and address behavioural disorders, ultimately helping to maintain independence and enhance the quality of life for individuals with AD. Despite these benefits, it is important to note that these treatments are not curative; they primarily alleviate symptoms rather than address the underlying causes of AD.61
Memantine, a non-competitive NMDA receptor antagonist, has an unclear but likely mechanism of action. It is thought to mitigate the excitatory neurotoxic effects of glutamate by blocking ionotropic receptors, particularly since glutamate levels are pathologically elevated in AD.60,62 While AChE inhibitors have been clinically utilized for mild to moderate AD, there were no available treatments for patients with moderate to severe AD until 2003.59 The FDA subsequently approved memantine for moderate and severe AD, allowing it to be used either as a standalone therapy or in combination with AChE inhibitors.64 When used alone, memantine has shown modest efficacy and a favourable safety profile in treating moderate to severe AD. Additionally, combination therapy with AChE inhibitors appears to provide added benefits over monotherapy.60 However, similar to AChE inhibitors, memantine’s effectiveness is limited in duration, and it does not significantly affect long-term disease progression. Its impact on patients with mild to moderate AD is minimal, making it unsuitable for individuals with mild AD.63 In response to the common practice of co-prescribing memantine and donepezil, a combination therapy known as Namzaric is now available as a convenient once-daily capsule.59
Limitation of current treatment choices
Despite decades of research into various therapeutic strategies, no curative treatment for AD currently exists, making prevention the primary focus.61 Existing therapies, including AChE inhibitors and NMDA receptor antagonists, offer some symptomatic relief for cognitive function.59 However, it has been shown that none of these approved medications provide a true cure; they are purely palliative, and their effectiveness tends to decline over time.62 The most frequently reported adverse events associated with AChE inhibitors are gastrointestinal issues, such as nausea, vomiting, diarrhoea, and anorexia. Additionally, tacrine, another AChE inhibitor, was removed from the market due to concerns over hepatotoxicity. Furthermore, these medications do not prevent neuronal loss, brain atrophy, or the subsequent cognitive decline associated with the disease.60 Another significant limitation in the treatment of AD is the late-stage administration of these drugs. Early intervention is crucial, necessitating improved early-stage diagnoses that incorporate additional biomarkers while considering risk factors such as family history, genetic predisposition (e.g., ε4 allele), and memory complaints. Although current drug development efforts have concentrated on targeting amyloid plaques, there is a pressing need to explore new therapeutic targets.61
In response to these limitations, the development of disease-modifying therapies aimed at altering the progression of AD has become a global priority. However, despite significant efforts over the past few decades, no new medication has been approved by the FDA since memantine in 2003. In fact, most candidate drugs targeting Aβ have not demonstrated clinical efficacy in late-stage trials.60 This modest effectiveness can be attributed to the challenges associated with delivering drugs across the restrictive BBB, which has led to many trial failures. Moreover, higher dosages can increase the risk of side effects, and age-related changes in neuronal membranes and receptors may further reduce drug efficacy.61 The primary reasons for the failure of disease-modifying treatments for AD include the initiation of treatment too late in the disease progression, incorrect drug dosages, poor selection of treatment targets, and a fundamental lack of understanding of AD’s complex pathophysiology. This suggests that patients may remain unresponsive to monotherapy, indicating a need for combination therapies rather than single-agent treatments. Furthermore, multi-target drug strategies are necessary to address multiple pathways involved in the disease.65 In addition to pharmacological interventions, managing underlying conditions such as hyperlipidaemia, diabetes, and hypertension is crucial for AD patients. Careful monitoring of hydration, sleep, nutrition, and the treatment of deficiencies (including thyroid, electrolyte, vitamin B12, vitamin D, and folate) as well as systemic conditions (such as infections, pain, and constipation) is essential.64 Other challenges include low bioavailability, increased serum half-life, and the difficulty of crossing the BBB.66
Therapeutic interventions for managing AD
In recent years, pre-clinical and clinical studies on AD have significantly advanced our understanding of its underlying mechanisms and potential treatments. Pre-clinical research has concentrated on key factors such as Aβ plaques, tau protein accumulation, and neuroinflammation, contributing to the identification of new therapeutic targets. Meanwhile, clinical trials conducted during this period have explored various approaches, including monoclonal antibodies and gene therapies, with the aim of slowing disease progression and improving cognitive function in patients. These studies represent critical steps toward developing more effective treatments for AD. Below, we discuss some of the reported pre-clinical and clinical studies on AD conducted in the last five years (2018-2024).
The role of natural compounds in treating AD
Natural phytochemicals are promising in AD research due to their neuroprotective, antioxidant, and Aβ-inhibiting properties, with low toxicity, enhancing their therapeutic potential. For instance, resveratrol, a natural polyphenolic stilbene abundantly found in grapes, berries, peanuts, and red wine, protects PC-12 cells (a widely used rat pheochromocytoma cell line derived from the adrenal medulla and a classical in vitro model for neuronal differentiation, neurotransmitter release, and neurobiological signaling) by increasing mitophagy and related protein expression, effects blocked by 3‑methyladenine (3-MA), highlighting mitophagy as a key mechanism and a potential therapeutic target in AD.67 Similarly, apigenin, a natural compound found in fruits and vegetables, exhibits neuroprotective effects in lipopolysaccharide (LPS)-induced neuroinflammation in rat neuron–glia co-cultures. At 1 μM, it preserved neuronal and astrocytic integrity, reduced microglial activation, modulated cytokine expression, and increased brain‑derived neurotrophic factor (BDNF) levels, highlighting its potential as a neuro-immunomodulatory agent for AD.68
Tanshinone IIA, a compound derived from Salvia miltiorrhiza Bunge, has also been investigated for its efficacy in treating AD. In vivo study demonstrated that administration of Tanshinone IIA (20 mg/kg) to APP/PS1 transgenic mice harboring mutations associated with familial early-onset AD that lead to accelerated Aβ overproduction and amyloid plaque deposition significantly improved spatial learning and memory. These effects were attributed to reduced Aβ accumulation, preservation of synaptic integrity, decreased neuronal cell death, suppression of glial activation, and inhibition of the receptor for advanced glycation end products (RAGE/NF-κB) signaling pathway through downregulation of pro-inflammatory cytokines, highlighting Tanshinone IIA’s therapeutic potential in AD. 69 In a similar approach, Yang et al. studied sulforaphane, an active compound from Raphani semen, at doses of 25 and 50 mg/kg, to treat STZ-induced cognitive deficits in rats. Sulforaphane reduced Alzheimer’s-related symptoms by lowering TNF-α and IL-6 levels while increasing IL-10 expression. The study further revealed that sulforaphane inhibited tau phosphorylation, activated the PI3K/AKT/GSK-3β signaling pathway, and decreased nitric oxide and NF-kB activation, underscoring its neuroprotective role in mitigating Alzheimer’s pathology.70 In addition, astaxanthin, a xanthophyll carotenoid, has shown similarly promising results, particularly in enhancing memory and cognition. In Alzheimer’s models, a 28-day treatment with astaxanthin at 1 mg/kg led to notable reductions in hippocampal soluble Aβ (1-42) levels, IRS-1 Ser307 phosphorylation (an insulin resistance marker), glycogen synthase kinase‑3β (GSK-3β) activity, TNF-α levels, AChE activity, nitrite levels, and oxidative stress. These findings suggest that astaxanthin not only has anti-amyloidogenic and neuroprotective properties but may also play a beneficial role in improving insulin resistance associated with AD.71
Cinnamic acid, a naturally occurring plant-derived compound, has shown promise in Alzheimer’s models by reducing cerebral Aβ plaque burden and improving cognitive function in animals through the stimulation of lysosomal biogenesis.72 Building on this, anthocyanins derived from Korean black beans have also demonstrated antioxidant and neuroprotective effects in Alzheimer’s models. In a study by Ali et al. (2018), anthocyanins were found to modulate the PI3K/Akt/GSK3β signalling pathway, reducing oxidative stress and apoptosis markers induced by Aβ. These effects were further supported by activation of the Nrf2/HO-1 antioxidant pathway, which led to improvements in cognitive markers in APP/PS1 mice, highlighting anthocyanin’s potential as a dietary neuroprotective supplement for AD.73 Likewise, rosmarinic acid has been found to reduce Aβ accumulation through distinct mechanisms, such as enhancing dopamine signalling and increasing levels of monoamines like dopamine and norepinephrine in the cerebral cortex, while downregulating monoamine oxidase B in key brain regions. This elevation in monoamines was shown to inhibit Aβ aggregation, suggesting that rosmarinic acid’s modulation of dopamine and monoamine levels may provide protective effects against Alzheimer’s.74 Extending these findings, sodium rutin has shown efficacy in enhancing Aβ clearance by upregulating phagocytosis-related receptors in microglia and promoting a metabolic shift from anaerobic glycolysis to mitochondrial oxidative phosphorylation (OXPHOS), which supplies the energy necessary for effective Aβ clearance. This shift resulted in reduced neuroinflammation, improved mitochondrial function, and enhanced synaptic plasticity, collectively reversing deficits in spatial learning and memory. These combined findings underscore sodium rutin’s potential as a therapeutic agent for AD.75
In a Tau-P301S mouse model, rutin treatment over 30 days was shown to reduce pathological tau, decrease tau hyperphosphorylation, mitigate gliosis and neuroinflammation, and enhance cognitive function. These beneficial effects were attributed to protein phosphatase 2A (PP2A) activation and downregulation of the NF-kB pathway, highlighting rutin’s dual potential in targeting both tau and Aβ pathologies in AD.76 Complementing these findings, another study demonstrated that jujuboside A (JuA) upregulates HSP90β via the Axl/ERK pathway, which helps maintain peroxisome proliferator‑activated receptor gamma (PPARγ) levels and promotes Aβ42 clearance. JuA treatment improved cognitive function and reduced Aβ42 plaque levels in APP/PS1 mice, though these effects were nullified when an Axl inhibitor was introduced, underscoring the importance of Axl signaling in JuA’s mechanism of action and potential as an AD therapy.77 Similarly, Qingxin kaiqiao Fang, a traditional Chinese medicine, was evaluated for its efficacy in AD by modulating the PI3K/AKT/GSK3β signalling pathway. This study identified 295 chemical components within the formulation, which collectively enhanced spatial cognition, learning, and memory in APP/PS1 mice. Qingxin kaiqiao Fang also protected PC-12 cells from Aβ25-35-induced apoptosis and reduced tau hyperphosphorylation, likely through its influence on the PI3K/Akt/GSK3β pathway, further supporting its therapeutic potential in AD.78
The role of the nanodelivery system improves AD
Nanodelivery systems offer targeted AD therapy by enhancing drug bioavailability, crossing the BBB, and delivering therapeutic agents directly to the brain. They enable sustained release, prolong effects, and minimize side effects, making them a promising approach for the precise and effective treatment of neurodegenerative disorders. A study by Cano et al. (2019), focused on developing a dual drug delivery system that incorporated Epigallocatechin-3-gallate (EGCG) and ascorbic acid, both loaded into PEGylated PLGA nanoparticles (EGCG/AA NPs), to enhance the stability of EGCG during delivery. The results showed that these nanoparticles improved the stability of EGCG in the brain after release compared to pure EGCG. Treatment with these nanoparticles in APP/PS1 mice led to a significant increase in synaptic density, a reduction in neuronal cell inflammation, and a decrease in Aβ plaques, ultimately contributing to improvements in memory and spatial learning.79 Similarly, a team of researchers developed a mesoporous nano-selenium (MSe) delivery system containing borneol and β-cyclodextrin nanovalves (Fc-β-CD), loaded with resveratrol (MSe-Res/Fc-β-CD/Bor) to target AD. This study demonstrated that the delivery system facilitated the release of borneol via esterase interaction, enabling effective BBB penetration. The subsequent redox-responsive release of resveratrol at the lesion site significantly reduced Aβ aggregation, oxidative stress, and tau hyperphosphorylation, improved memory in APP/PS1 mice, and enhanced the pharmacokinetic profile.80 In contrast, a different approach involved the use of siRNA nanocarriers made from PEGylated poly(2-(N,N-dimethylamino)ethyl methacrylate) (PEG-PDMAEMA), further modified with CGN and Tet1 peptides, to target β-site amyloid precursor protein cleaving enzyme 1 (BACE1) for Alzheimer’s management. Experimental results showed that these nanocarriers effectively penetrated the BBB, delivered the siRNA specifically to neurons via clathrin-mediated endocytosis, and reduced BACE1 mRNA levels by 50%, reversing amyloid-induced synaptic damage. In vivo studies demonstrated a significant decrease in BACE1 expression, a reduction in amyloid plaques, and improved cognitive function in APP/PS1 transgenic mice, without any observed side effects. These findings highlight the potential of these advanced nanocarriers for AD treatment.81
Inflammatory responses, characterized by oxidative stress and glial cell activation, have been recognized as key factors in the early stages of AD. In response to the interplay between inflammation and brain cells, Liu et al. (2021) developed a ROS-responsive dendrimer-peptide conjugate (APBP) designed to target the AD microenvironment and inhibit early-stage inflammatory responses. Their study demonstrated a significant reduction in ROS levels, a decrease in Aβ burden, normalized glial cell activity, and improved cognitive function in APPswe/PSEN1dE9 model mice, positioning this conjugate system as a promising multi-target therapeutics for AD.82 In a related study, Gao et al. (2020) designed T807/TPP-RBC-NPs nanoparticles to deliver antioxidants to neuronal mitochondria. By loading antioxidants into red blood cell membrane-camouflaged human serum albumin nanoparticles, which bore T807 and triphenylphosphine (TPP) molecules, these nanoparticles were shown to have enhanced stability, biocompatibility, and long-term circulation, while effectively penetrating the BBB and localizing in nerve cell mitochondria. When curcumin was encapsulated in these nanoparticles and administered to male Institute of Cancer Research (ICR) mice at a dose of 5 mg/kg, a notable reduction in mitochondrial oxidative stress and neuronal death was observed, underscoring their therapeutic potential.83 Similarly, another study developed a brain-targeted nanoparticle, PLGA-PEG-B6, loaded with curcumin to overcome its hydrophobicity and low bioavailability. In vitro tests revealed improved cellular uptake and blood compatibility of the nanoparticle, and in APP/PS1 transgenic mice, PLGA-PEG-B6/Cur significantly enhanced spatial learning and memory, while reducing hippocampal Aβ deposits and tau hyperphosphorylation, in contrast to native curcumin. These findings further highlight the potential of PLGA-PEG-B6/Cur nanoparticles as an effective treatment for AD.84
In addition, another study demonstrated that encapsulating quercetin in solid lipid nanoparticles and nanostructured lipid carriers enhanced their permeability to cross the BBB. These nanoparticles exhibited no cytotoxicity on human cerebral microvascular endothelial cell line (hCMEC/D3) cells when treated at 30 μM for 4 h, indicating their safety for use in brain-targeted therapies.85 Similarly, research focused on vitamin D-binding protein (DBP) encapsulated in PLGA nanoparticles showed significant inhibition of Aβ aggregation at a dose of 2.5 mg/kg. When delivered intravenously to 5XFAD mice, these nanoparticles notably reduced Aβ accumulation, neuroinflammation, neuronal loss, and cognitive dysfunction, suggesting that encapsulating DBP in nanoparticles could be a promising strategy for Alzheimer’s treatment.86 In another study, Meng et al. (2018) developed Huperzine A (HupA)-loaded lactoferrin-conjugated N-trimethylated chitosan (Lf-TMC) PLGA nanoparticles for intranasal delivery to the brain in Alzheimer’s therapy. These nanoparticles demonstrated high drug entrapment efficiency, sustained release over 48 h, and significantly enhanced mucin adsorption and reduced cytotoxicity compared to non-targeted nanoparticles. In vivo imaging confirmed improved brain targeting and drug accumulation in key regions, highlighting the potential of Lf-TMC nanoparticles for targeted intranasal HupA delivery in Alzheimer’s therapy.87 Furthermore, Hou et al. (2020) investigated the use of chiral gold nanoparticles as a therapeutic strategy to prevent Aβ aggregation in AD. They synthesized L- and D-glutathione-stabilized gold nanoparticles, with D3.3 showing higher binding affinity for Aβ42 and better brain distribution than L3.3. D3.3 effectively inhibited Aβ42 fibrillation and improved behavioral outcomes in AD model mice, indicating that chiral gold nanoparticles, like D3.3, may provide a promising approach for targeting Aβ aggregation in AD.88
A study conducted in 2018 revealed that nicotinamide-loaded solid lipid nanoparticles (SLNs) functionalized with polysorbate 80 (S80), phosphatidylserine (PS), or phosphatidic acid were generally safe, with the exception of the S80-functionalized SLNs. These nanoparticles showed improved biodistribution to brain tissue, confirming their effectiveness in brain delivery and treatment of AD. Notably, the PS-functionalized SLNs demonstrated enhanced cognition, preserved neurons, and reduced tau hyperphosphorylation more effectively than conventional oral nicotinamide when administered via intraperitoneal injection to rats, highlighting their potential in enhancing brain delivery systems for early AD treatment.89 In a different approach, Jiang and colleagues designed a novel dual-target inhibitor for GSK-3β and AChE, with compound 2f emerging as the most promising among the synthesized inhibitors. This compound exhibited potent inhibition of both hAChE and hGSK-3β, with IC50 values of 6.5 nM and 66 nM, respectively. Treatment with compound 2f resulted in a 46% reduction in Aβ self-aggregation and inhibition of tau hyperphosphorylation in N2a-Tau cells. This study demonstrated improved cognitive function in scopolamine-treated mice and reduced hepatotoxicity compared to tacrine, suggesting its potential as a dual-target treatment for AD.90 Similarly, Najafi et al. (2019) synthesized tacrine-coumarin hybrids linked to 1,2,3-triazole, with compounds 8e and 8m showing potent anti-acetylcholinesterase and anti-butyrylcholinesterase activities, with IC50 values of 27 nM and 6 nM, respectively, outperforming tacrine and donepezil. While compound 8e exhibited low BACE1 inhibitory activity and neuroprotection in PC-12 cells, it significantly reversed scopolamine-induced memory deficits in rats, as evidenced by the Morris water maze test.91
In a study by Abozaid et al. (2022), resveratrol-selenium nanoparticles (RSV-SeNPs) were evaluated for their neuroprotective effects in an Alzheimer’s model induced by aluminum chloride in rats. The RSV-SeNPs effectively reduced oxidative stress, improved cholinergic function, and prevented tau hyperphosphorylation by activating the PI3K pathway. Additionally, they decreased neuroinflammation and promoted neurite outgrowth through modulation of SIRT1 and microRNA-134. These findings suggest that RSV-SeNPs could be a promising therapeutic for AD, enhancing antioxidant and anti-inflammatory responses to improve neurocognitive function.92 In contrast, another study developed a glycosylated polymeric nanomedicine (Gal-NP@siRNA) for targeted delivery of siRNA that silences BACE1 in an AD model. This nanomedicine reduced Aβ aggregation, a hallmark of AD, and showed stability in the bloodstream while crossing the BBB via Glut1-mediated transport. In the APP/PS1 mouse model, Gal-NP@siRNA significantly reduced BACE1 expression, improved cognitive function, and demonstrated no adverse effects, offering a promising strategy for RNA interference in neurodegenerative diseases like AD.93 Similarly, a study by Wang et al. investigated a novel series of quinoline-indole derivatives, which showed multiple beneficial effects, including antioxidant properties, BBB penetration, biometal chelation, modulation of Aβ aggregation, and neuroprotection in a mouse model. Among these derivatives, compound 8d (WI-1758) exhibited hippocampal cell proliferation, good liver metabolic stability, and favourable pharmacokinetics, with an oral bioavailability of 14.1% and a positive log BB (-0.19), indicating its potential to cross the BBB. Chronic oral administration of 8d·HCl significantly enhanced cognitive and spatial memory in APP/PS1 AD mice while reducing cerebral Aβ deposits, suggesting that compound 8d could be a promising therapeutic candidate for AD due to its ability to penetrate the BBB, decrease Aβ deposits, and improve cognitive function in mouse models.94
In addition, a series of novel nanocomposites (CeNC/IONC/MSN-T807) loaded with methylene blue, a tau aggregation inhibitor, was developed. These nanocomposites demonstrated a high binding affinity to hyperphosphorylated tau and effectively inhibited multiple pathways involved in tau-related AD pathology. Treatment with these nanocomposites resulted in improved memory, reduced mitochondrial oxidative stress, suppressed tau hyperphosphorylation, and prevented neuronal death both in vitro and in vivo, highlighting their potential as a therapeutic intervention for managing AD.95 Similarly, Arslan and colleagues explored the neuroprotective effects of farnesene sesquiterpene, which showed promise in reducing Aβ toxicity in human neuroblastoma (SHSY-5Y) cells. Their study revealed that farnesene sesquiterpene significantly improved cell viability, reduced necrotic cell death, enhanced antioxidant capacity, and lowered oxidative stress, positioning it as a potential neuroprotective and anti-necrotic agent for treating neurodegenerative disorders.96 In a related study, the inhibitory effects of metformin on tau hyperphosphorylation in diabetic encephalopathy (DE) were investigated. The results demonstrated that metformin treatment in db/db mice significantly improved cognitive function, reduced tau hyperphosphorylation, and restored autophagy. In addition, treatment with metformin in HT22 cells cultured under hyperglycemic conditions enhanced autophagy clearance and mitigated the risk of diabetes-induced tau hyperphosphorylation.97 Furthermore, Zhao et al. developed a novel nanocomposite (14 ± 4 nm) that effectively eliminated toxic Aβ aggregates and reduced Aβ-induced neurotoxicity in AD mice. Their findings suggested that these nanocomposites altered the structure of Aβ aggregates, transforming them into Aβ/nanocomposite nanoclusters instead of harmful Aβ oligomers. These nanocomposites also reduced the pathological oligomers, minimized neuronal damage, restored the ability of microglia to clear Aβ, and protected hippocampal neurons from apoptosis.98 Collectively, these studies indicate that small-sized nanocomposites, as well as other neuroprotective agents, could provide promising approaches for developing new treatments for AD.
During AD, disruption of the BBB increases its permeability, allowing harmful molecules to enter the brain and accelerating neurodegeneration. Consequently, developing therapies that can effectively cross the BBB is crucial for effective Alzheimer’s treatment. For example, a leading group of researchers developed memantine-loaded nanoparticles (MEM-PEG-PLGA NPs) with a mean particle size of 152.6 nm. These nanoparticles improved drug delivery in Alzheimer’s by reducing Aβ plaques, crossing the BBB, and enhancing memory function in transgenic mice.99 Similarly, Han and colleagues designed a genistein-encapsulated delivery system (RVG/TPP-MASLNs-GS), which efficiently evaded immune clearance, crossed the BBB, and delivered antioxidants to mitochondria, promoting uptake in HT-22 cells.100 These studies highlight the significance of nanoparticle-based systems in overcoming the BBB and enhancing drug efficacy in Alzheimer’s treatment. In contrast, another study developed a series of tacrine-tryptophan heterodimers for AD, with the compound S-K1035 showing strong inhibitory activity against human acetylcholinesterase and butyrylcholinesterase, with IC50 values of 6.3 nM and 9.1 nM, respectively. This compound also demonstrated inhibitory effects on Aβ42 self-aggregation and hAChE-induced Aβ40 aggregation while crossing the BBB and inhibiting neuronal nitric oxide synthase enzyme activity. These findings further underscore the potential of multi-target agents like these heterodimers in managing AD, complementing the strategies developed in the previous studies for effective drug delivery and brain penetration.79
Ongoing clinical trials for AD
Ongoing AD clinical trials focus on disease-modifying therapies targeting key pathological mechanisms rather than symptoms. Anti-amyloid agents, including monoclonal antibodies like aducanumab and lecanemab, aim to reduce Aβ plaques, though their long-term efficacy remains debated. Tau-targeting therapies, such as gosuranemab and tilavonemab, seek to prevent tau aggregation, with limited clinical impact so far. Approaches addressing neuroinflammation, neuroprotection, and mitochondrial dysfunction are also under investigation, alongside regenerative strategies like stem cell therapy to restore neuronal function. These trials, at varying recruitment stages, reflect the multifactorial nature of AD and are summarized in Tables 2-4.
Table 2: Key active ad clinical trials (not recruiting) with interventions and duration
| NCT number | Intervention | Start date | Completion date |
| NCT05891496 | Semaglutide | 6/20/2023 | 9/11/2025 |
| NCT05269394 | Lecanemab | 12/22/2021 | 2028-07 |
| NCT06292351 | DMB-I (Dimebon) | 12/27/2023 | 12/21/2024 |
| NCT05476783 | TB006 | 9/14/2022 | 2024-10 |
| NCT04867616 | Bepranemab | 6/9/2021 | 7/28/2025 |
| NCT05352763 | Simufilam | 5/12/2022 | 11/29/2025 |
| NCT05267535 | Piromelatine | 5/12/2022 | 2025-06 |
| NCT05310071 | Aducanumab | 6/2/2022 | 10/31/2026 |
| NCT03634007 | LX1001 | 11/6/2019 | 2024-11 |
| NCT05771428 | ABBV-552 | 4/27/2023 | 9/5/2024 |
Table 3: Key currently recruiting clinical trials for AD interventions
| NCT Number | Intervention | Start date | Completion date |
| NCT04639050 | RO7126209 | 3/15/2021 | 12/31/2028 |
| NCT01760005 | Gantenerumab, solanezumab, lecanemab | 2012-12 | 2028-07 |
| NCT02833792 | Human mesenchymal stem cells & lactated Riunger’s solution | 6/1/2016 | 12/31/2024 |
| NCT04408755 | AVP-786 | 7/8/2020 | 3/31/2026 |
| NCT04464564 | AVP-786 | 9/3/2020 | 11/30/2026 |
| NCT02446132 | AVP-786 | 2015-12 | 1/30/2025 |
| NCT04308512 | Care4AD system | 11/1/2021 | 12/1/2025 |
| NCT05811442 | 50561 | 4/18/2023 | 2024-06 |
| NCT06079190 | GSK4527226 | 10/20/2023 | 5/16/2029 |
| NCT05804383 | BMS-984923 | 3/28/2023 | 7/15/2025 |
Table 4: Key AD clinical trials not yet recruiting/ enrolling by invitation
| NCT number | Intervention | Start date | Completion date |
| NCT06194799 | ACP-204 | 4/23/2024 | 2029-05 |
| NCT06159673 | ACP-204 | 11/14/2023 | 2028-02 |
| NCT06427668 | SPG302 | 2024-07 | 2026-06 |
| NCT06514066 | Cannabidiol oil | 9/1/2024 | 10/1/2025 |
| NCT06135051 | Diadem prototype | 2024-04 | 2027-04 |
| NCT05606341 | CpG1018 | 3/13/2023 | 2025-11 |
| NCT06247345 | ADEL-Y01 | 2/5/2024 | 5/31/2026 |
| NCT06234436 | Plasma exchange | 12/7/2023 | 12/31/2025 |
| NCT06022068 | Sirolimus | 9/1/2023 | 1/31/2025 |
| NCT06120049 | [18F]-MFBG PET CT / PET MRI | 2023-12 | 2026-12 |
Future perspective
The future of AD treatment depends on overcoming major challenges in CNS drug delivery, as most current therapies remain largely symptomatic. The BBB and brain complexity limit effective drug penetration, underscoring the need for innovative delivery strategies to enhance bioavailability. Multidisciplinary and precision-medicine approaches integrating genetics, neuroscience, and immunology may enable personalized, disease-modifying therapies. However, translating these approaches into clinically effective treatments remains slow and resource-intensive. Early diagnosis and preclinical detection will be critical for timely intervention, alongside greater emphasis on prevention and risk-factor modification, though the implementation of large-scale screening strategies poses ethical, economic, and technical challenges. Novel systems such as nanoparticles, exosomes, and stem cell therapies show promise but face challenges related to safety, scalability, regulation, and sustained efficacy, which currently limit their widespread clinical application. Overall, meaningful progress will require integrated strategies focusing on prevention, early intervention, improved delivery, and truly disease-modifying treatments, supported by rigorous clinical validation and long-term outcome studies.
Conclusion
In conclusion, while significant progress has been made in understanding the molecular mechanisms of AD, key challenges remain. The complex interplay between Aβ aggregation, tau hyperphosphorylation, neuroinflammation, oxidative stress, and mitochondrial dysfunction drives the disease’s progression. However, advancements in molecular biology, genetics, and neuroscience have opened new therapeutic avenues, such as targeted modulation of amyloid and tau, immune regulation, and epigenetic approaches. Innovative drug delivery systems, like nanoparticles, offer promise in overcoming the BBB, potentially improving treatment efficacy. Nevertheless, issues related to safety, reproducibility, and clinical translatability continue to hinder their successful advancement. Despite these advancements, the translation of these findings into effective clinical therapies remains a challenge, particularly with AD’s complex, multifactorial nature. The focus on early detection and personalized medicine holds great potential for more effective interventions, although robust biomarkers and validated clinical endpoints are still required. As research continues to elucidate the complexities of AD, measured expectations are warranted, as meaningful therapeutic breakthroughs will depend on sustained translational research, rigorous clinical trials, and effective regulatory alignment.
Acknowledgement
The authors sincerely thank Maharaja Agrasen University, Himachal Pradesh; Panjab University, Chandigarh; CGC University, Punjab; and Saraswati College of Pharmacy, Punjab, India, for providing the space, and time to enable this collaborative work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable
Author contributions
- Shivani Thakur: Literature review, data collection and drafting the manuscript
- Pankaj Kumar: Analysis, interpretation, and editing
- Sourbh Suren Garg: Review writing and analysis
- Ankush Goyal: Review writing
- Pradeep Goyal: Analysis and editing
- Mona Piplani: Supervision, editing and final approval of the manuscript
References
- Calabrò M, Rinaldi C, Santoro G, Crisafulli C. The biological pathways of Alzheimer disease: a review. AIMS Neurosci. 2020;8(1):86‑132.
CrossRef - Greene AN, Solomon MB, Privette Vinnedge LM. Novel molecular mechanisms in Alzheimer’s disease: the potential role of DEK in disease pathogenesis. Front Aging Neurosci. 2022;14:1018180.
CrossRef - Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6(11):1054‑1061.
CrossRef - Iqbal K, Wiśniewski HM, Shelanski ML, Brostoff S, Liwnicz BH, Terry RD. Protein changes in senile dementia. Brain Res. 1974;77(2):337‑343.
CrossRef - Ma N, Tie C, Yu B, Zhang W, Wan J. Identifying lncRNA‑miRNA‑mRNA networks to investigate Alzheimer’s disease pathogenesis and therapy strategy. Aging (Albany NY). 2020;12(3):2897‑2913.
CrossRef - Ossenkoppele R, van der Kant R, Hansson O. Tau biomarkers in Alzheimer’s disease: towards implementation in clinical practice and trials. Lancet Neurol. 2022;21(8):726‑734.
CrossRef - El‑Hussieny M, Abd‑El‑Maksoud MA, Soliman FM, Fouad MA, El‑Ashrey MK. Dual‑target ligand discovery for Alzheimer’s disease: triphenylphosphoranylidene derivatives as inhibitors of acetylcholinesterase and β‑amyloid aggregation. J Enzyme Inhib Med Chem. 2023;38(1):2166040.
CrossRef - Butterfield DA. Amyloid β‑peptide (1‑42)‑induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain: a review. Free Radic Res. 2002;36(12):1307‑1313.
CrossRef - Uddin MS, Kabir MT, Niaz K, et al. Molecular insight into the therapeutic promise of flavonoids against Alzheimer’s disease. 2020;25(6):1267.
CrossRef - Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2013;6(1):19‑33.
CrossRef - Breijyeh Z, Karaman R. Comprehensive review on Alzheimer’s disease: causes and treatment. 2020;25(24):5789.
CrossRef - Rajmohan R, Reddy PH. Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J Alzheimers Dis. 2017;57(4):975‑999.
CrossRef - Mohamed Asik R, Suganthy N, Aarifa MA, et al. Alzheimer’s disease: a molecular view of β-amyloid induced morbific events. 2021;9(9):1126.
CrossRef - Brandl S, Reindl M. Blood-brain barrier breakdown in neuroinflammation: current in vitro models. Int J Mol Sci. 2023;24(16):12699.
CrossRef - Zhang H, Jiang X, Ma L, et al. Role of Aβ in Alzheimer’s-related synaptic dysfunction. Front Cell Dev Biol. 2022;10:964075.
CrossRef - Cai Q, Tammineni P. Mitochondrial aspects of synaptic dysfunction in Alzheimer’s disease. J Alzheimers Dis. 2017;57(4):1087‑1103.
CrossRef - Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8(21):2003‑2014.
CrossRef - Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23(4):213‑227.
CrossRef - Yang KF, Zhang JY, Feng M, et al. Secretase promotes AD progression: simultaneously cleave Notch and APP. Front Aging Neurosci. 2024;16:1445470.
CrossRef - Huang X, Fowler C, Li Y, et al. Clearance and transport of amyloid-β by peripheral monocytes correlate with Alzheimer’s disease progression. Nat Commun. 2024 Sep 12;15(1):7998.
CrossRef - Walter J. Control of amyloid-β-peptide generation by subcellular trafficking of the β-amyloid precursor protein and β-secretase. Neurodegener Dis. 2006;3(4‑5):247‑254.
CrossRef - Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2(5):a006270.
CrossRef - Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189.
CrossRef - Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595‑608.
CrossRef - O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34(1):185‑204.
CrossRef - Zhang C, Browne A, DiVito JR, et al. Amyloid-β production via cleavage of amyloid-β protein precursor is modulated by cell density. J Alzheimers Dis. 2010;22(2):683‑694.
CrossRef - Bharadwaj PR, Dubey AK, Masters CL, Martins RN, Macreadie IG. Aβ aggregation and possible implications in Alzheimer’s disease pathogenesis. J Cell Mol Med. 2009;13(3):412‑421.
CrossRef - Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388‑405.
CrossRef - Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2(7):a006338.
CrossRef - Hampel H, Hardy J, Blennow K, et al. The amyloid-β pathway in Alzheimer’s disease. Mol Psychiatry. 2021;26(10):5481‑5503.
CrossRef - Iqbal K, Alonso AD, Chen S, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta Mol Basis Dis. 2005;1739(2‑3):198‑210.
CrossRef - Medeiros R, Baglietto‑Vargas D, LaFerla FM. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci Ther. 2011;17(5):514‑524.
CrossRef - Otero‑Garcia M, Mahajani SU, Wakhloo D, et al. Molecular signatures underlying neurofibrillary tangle susceptibility in Alzheimer’s disease. 2022;110(18):2929‑2948.
CrossRef - Walker LC, Lynn DG, Chernoff YO. A standard model of Alzheimer’s disease? 2018;12(5‑6):261‑265.
CrossRef - Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. 2017;140(12):3286‑3300.
CrossRef - Kocahan S, Doğan Z. Mechanisms of Alzheimer’s disease pathogenesis and prevention: the brain, neural pathology, N-methyl-D-aspartate receptors, tau protein and other risk factors. Clin Psychopharmacol Neurosci. 2017;15(1):1‑14.
CrossRef - Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013;70(3):304‑310.
CrossRef - Mandrekar‑Colucci S, Landreth GE. Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):156‑167.
CrossRef - Choi SB, Kwon S, Kim JH, et al. The molecular mechanisms of neuroinflammation in Alzheimer’s disease, the consequence of neural cell death. Int J Mol Sci. 2023;24(14):11757.
CrossRef - Morales I, Guzmán‑Martínez L, Cerda‑Troncoso C, Farías GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer’s disease: a rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112.
CrossRef - Cai Y, Liu J, Wang B, Sun M, Yang H. Microglia in the neuroinflammatory pathogenesis of Alzheimer’s disease and related therapeutic targets. Front Immunol. 2022;13:856376.
CrossRef - Rani V, Verma R, Kumar K, Chawla R. Role of pro-inflammatory cytokines in Alzheimer’s disease and neuroprotective effects of pegylated self-assembled nanoscaffolds. Curr Res Pharmacol Drug Discov. 2023;4:100149.
CrossRef - Dhapola R, Hota SS, Sarma P, et al. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. 2021;29(6):1669‑1681.
CrossRef - Walker KA, Ficek BN, Westbrook R. Understanding the role of systemic inflammation in Alzheimer’s disease. ACS Chem Neurosci. 2019;10(8):3340‑3342.
CrossRef - Ní Chasaide C, Lynch MA. The role of the immune system in driving neuroinflammation. J Neuroinflammation. 2020;4:2398212819901082.
CrossRef - Liu C, Xu S, Liu Q, et al. Identification of immune cells infiltrating in hippocampus and key genes associated with Alzheimer’s disease. BMC Med Genomics. 2023;16(1):53.
CrossRef - Dai L, Shen Y. Insights into T-cell dysfunction in Alzheimer’s disease. Aging Cell. 2021;20(12):e13511.
CrossRef - Babić Leko M, Nikolac Perković M, Klepac N, et al. IL-1β, IL-6, IL-10, and TNF-α single nucleotide polymorphisms influence susceptibility to Alzheimer’s disease pathology. J Alzheimers Dis. 2020 Jun 2;75(3):1029-1047.
CrossRef - Juárez-Cedillo T, Martínez-Rodríguez N, Vargas-Alarcón G, et al. Synergistic influence of cytokine gene polymorphisms on the risk of dementia: a multifactor dimensionality reduction analysis. Front Aging Neurosci. 2022 Oct 26;14:952173.
CrossRef - Huang WJ, Zhang X, Chen WW. Role of oxidative stress in Alzheimer’s disease. Biomed Res. 2016;4(5):519‑522.
CrossRef - Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 2014;1842(8):1240‑1247.
CrossRef - Wang W, Zhao F, Ma X, Perry G, Zhu XM. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. 2020;15(1):30.
CrossRef - Troutwine BR, Strope TA, Franczak E, et al. Mitochondrial function and Aβ in Alzheimer’s disease postmortem brain. Neurobiol Dis. 2022 Sep 1;171:105781.
CrossRef - Misrani A, Tabassum S, Yang LM. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front Aging Neurosci. 2021;13:617588.
CrossRef - Sultana MA, Hia RA, Akinsiku O, Hegde V. Peripheral mitochondrial dysfunction: a potential contributor to the development of metabolic disorders and Alzheimer’s disease. 2023;12(7):1019.
CrossRef - Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta Mol Basis Dis. 2014;1842(8):1219‑1231.
CrossRef - Lopez Sanchez MIG, van Wijngaarden P, Trounce IA. Amyloid precursor protein-mediated mitochondrial regulation and Alzheimer’s disease. Br J Pharmacol. 2019;176(18):3464‑3474.
CrossRef - Singh A, Faccenda D, Campanella M. Pharmacological advances in mitochondrial therapy. Essays Biochem. 2021;65:153‑169.
CrossRef - Fish PV, Steadman D, Bayle ED, Whiting P. New approaches for the treatment of Alzheimer’s disease. Bioorg Med Chem Lett. 2019;29(2):125‑133.
CrossRef - Vaz M, Silvestre S. Alzheimer’s disease: recent treatment strategies. Eur J Pharmacol. 2020;887:173554.
CrossRef - Passeri E, Elkhoury K, Morsink M, et al. Alzheimer’s disease: treatment strategies and their limitations. Int J Mol Sci. 2022;23(22):13954.
CrossRef - Folch J, Ettcheto M, Petrov D, et al. Review of the advances in treatment for Alzheimer disease: strategies for combating β-amyloid protein. Neurologia (Engl Ed). 2018;33(1):47‑58.
CrossRef - Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. 2019;179(2):312‑339.
CrossRef - Yiannopoulou KG, Papageorgiou SG. Current and future treatments in Alzheimer disease: an update. J Clin Neurosci Sci Dis. 2020;12:1179573520907397.
CrossRef - Yiannopoulou KG, Anastasiou AI, Zachariou V, Pelidou SH. Reasons for failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. 2019;7(4):97.
CrossRef - Srivastava S, Ahmad R, Khare SK. Alzheimer’s disease and its treatment by different approaches: a review. Eur J Med Chem. 2021;216:113320.
CrossRef - Wang H, Jiang T, Li W, Gao NA, Zhang T. Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol Lett. 2018;282:100‑108.
CrossRef - Dourado NS, Souza CD, De Almeida MM, et al. Neuroimmunomodulatory and neuroprotective effects of the flavonoid apigenin in in vitro models of neuroinflammation associated with Alzheimer’s disease. Front Aging Neurosci. 2020;12:119.
CrossRef - Ding B, Lin C, Liu Q, et al. Tanshinone IIA attenuates neuroinflammation via inhibiting RAGE/NF-κB signaling pathway in vivo and in vitro. J Neuroinflammation. 2020;17(1):302.
CrossRef - Yang W, Liu Y, Xu QQ, Xian YF, Lin ZX. Sulforaphene ameliorates neuroinflammation and hyperphosphorylated tau protein via regulating the PI3K/Akt/GSK‑3β pathway in experimental models of Alzheimer’s disease. Oxid Med Cell Longev. 2020;2020:4754195.
CrossRef - Rahman SO, Panda BP, Parvez S, et al. Neuroprotective role of astaxanthin in hippocampal insulin resistance induced by Aβ peptides in animal model of Alzheimer’s disease. Biomed Pharmacother. 2019;110:47‑58.
CrossRef - Chandra S, Roy A, Jana M, Pahan K. Cinnamic acid activates PPARα to stimulate lysosomal biogenesis and lower amyloid plaque pathology in an Alzheimer’s disease mouse model. Neurobiol Dis. 2019;124:379‑395.
CrossRef - Ali T, Kim T, Rehman SU, et al. Natural dietary supplementation of anthocyanins via PI3K/Akt/Nrf2/HO-1 pathways mitigate oxidative stress, neurodegeneration, and memory impairment in a mouse model of Alzheimer’s disease. Mol Neurobiol. 2018;55(7):6076‑6093.
CrossRef - Hase T, Shishido S, Yamamoto S, et al. Rosmarinic acid suppresses Alzheimer’s disease development by reducing amyloid β aggregation by increasing monoamine secretion. Sci Rep. 2019;9(1):8711.
CrossRef - Pan RY, Ma J, Kong XX, et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci Adv. 2019;5(2):eaau6328.
CrossRef - Sun XY, Li LJ, Dong QX, et al. Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2021;18(1):131.
CrossRef - Zhang M, Qian C, Zheng ZG, et al. Jujuboside A promotes Aβ clearance and ameliorates cognitive deficiency in Alzheimer’s disease through activating Axl/HSP90/PPARγ pathway. 2018;8(15):4262‑4276.
CrossRef - Liu S, Xu L, Shen Y, et al. Qingxin Kaiqiao Fang decreases Tau hyperphosphorylation in Alzheimer’s disease via the PI3K/Akt/GSK3β pathway in vitro and in vivo. J Ethnopharmacol. 2024;318:117031.
CrossRef - Cano A, Ettcheto M, Chang JH, et al. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s disease mice model. J Control Release. 2019;301:62‑75.
CrossRef - Sun J, Wei C, Liu Y, et al. Progressive release of mesoporous nano-selenium delivery system for the multi-channel synergistic treatment of Alzheimer’s disease. 2019;197:417‑431.
CrossRef - Wang P, Zheng X, Guo Q, et al. Systemic delivery of BACE1 siRNA through neuron-targeted nanocomplexes for treatment of Alzheimer’s disease. J Control Release. 2018;279:220‑233.
CrossRef - Liu P, Zhang T, Chen Q, et al. Biomimetic dendrimer-peptide conjugates for early multi-target therapy of Alzheimer’s disease by inflammatory microenvironment modulation. Adv Mater. 2021;33(26):2100746.
CrossRef - Gao C, Wang Y, Sun J, et al. Neuronal mitochondria-targeted delivery of curcumin by biomimetic engineered nanosystems in Alzheimer’s disease mice. Acta Biomater. 2020;108:285‑299.
CrossRef - Fan S, Zheng Y, Liu X, et al. Curcumin-loaded PLGA-PEG nanoparticles conjugated with B6 peptide for potential use in Alzheimer’s disease. Drug Deliv. 2018;25(1):1091‑1102.
CrossRef - Song JX, Malampati S, Zeng Y, et al. A small molecule transcription factor EB activator ameliorates beta‑amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell. 2020;19(2):e13069.
CrossRef - Jeon SG, Cha MY, Kim JI, et al. Vitamin D-binding protein-loaded PLGA nanoparticles suppress Alzheimer’s disease-related pathology in 5XFAD mice. Nanomedicine (Lond). 2019;17:297‑307.
CrossRef - Meng Q, Wang A, Hua H, et al. Intranasal delivery of Huperzine A to the brain using lactoferrin-conjugated N-trimethylated chitosan surface-modified PLGA nanoparticles for treatment of Alzheimer’s disease. Int J Nanomedicine. 2018;13:705‑718.
CrossRef - Hou K, Zhao J, Wang H, et al. Chiral gold nanoparticles enantioselectively rescue memory deficits in a mouse model of Alzheimer’s disease. Nat Commun. 2020;11(1):4790.
CrossRef - Vakilinezhad MA, Amini A, Akbari Javar H, et al. Nicotinamide loaded functionalized solid lipid nanoparticles improves cognition in Alzheimer’s disease animal model by reducing Tau hyperphosphorylation. DARU J Pharm Sci. 2018;26(2):165‑177.
CrossRef - Jiang XY, Chen TK, Zhou JT, et al. Dual GSK-3β/AChE inhibitors as a new strategy for multitargeting anti-Alzheimer’s disease drug discovery. ACS Med Chem Lett. 2018;9(3):171‑176.
CrossRef - Najafi Z, Mahdavi M, Saeedi M, et al. Novel tacrine-coumarin hybrids linked to 1,2,3-triazole as anti-Alzheimer’s compounds: In vitro and in vivo biological evaluation and docking study. Bioorg Chem. 2019;83:303‑316.
CrossRef - Abozaid OA, Sallam MW, El-Sonbaty S, et al. Resveratrol-selenium nanoparticles alleviate neuroinflammation and neurotoxicity in a rat model of Alzheimer’s disease by regulating Sirt1/miRNA-134/GSK3β expression. Biol Trace Elem Res. 2022;200(12):5104‑5114.
CrossRef - Zhou Y, Zhu F, Liu Y, et al. Blood-brain barrier-penetrating siRNA nanomedicine for Alzheimer’s disease therapy. Sci Adv. 2020;6(41):eabc7031.
CrossRef - Wang Z, Hu J, Yang X, et al. Design, synthesis, and evaluation of orally bioavailable quinoline-indole derivatives as innovative multitarget-directed ligands: Promotion of cell proliferation in the adult murine hippocampus for the treatment of Alzheimer’s disease. J Med Chem. 2018;61(5):1871‑1894.
CrossRef - Chen Q, Du Y, Zhang K, et al. Tau-targeted multifunctional nanocomposite for combinational therapy of Alzheimer’s disease. ACS Nano. 2018;12(2):1321‑1338.
CrossRef - Arslan ME, Türkez H, Mardinoğlu AJ. In vitro neuroprotective effects of farnesene sesquiterpene on Alzheimer’s disease model of differentiated neuroblastoma cell line. Int J Neurosci. 2021;131(8):745‑754.
CrossRef - Chen JL, Luo C, Pu D, et al. Metformin attenuates diabetes-induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagic clearance. Exp Neurol. 2019;311:44‑56.
CrossRef - Zhao Y, Cai J, Liu Z, et al. Nanocomposites inhibit the formation, mitigate the neurotoxicity, and facilitate the removal of β-amyloid aggregates in Alzheimer’s disease mice. Nano Lett. 2018;19(2):674‑683.
CrossRef - Sánchez-López E, Ettcheto M, Egea MA, et al. Memantine loaded PLGA PEGylated nanoparticles for Alzheimer’s disease: In vitro and in vivo characterization. J Nanobiotechnol. 2018;16(1):32.
CrossRef - Han Y, Gao C, Wang H, et al. Macrophage membrane-coated nanocarriers co-modified by RVG29 and TPP improve brain neuronal mitochondria-targeting and therapeutic efficacy in Alzheimer’s disease mice. Bioact Mater. 2021;6(2):529‑542.
CrossRef
Accepted on: 27-05-2026
Second Review by: Dr. Randa Salah Gomaa Mahmoud
Final Approval by: Dr. Gul Ozcan




