Mechanisms of Tumor Progression and Drug Resistance in Pancreatic Cancer Models (PANC-1, MiaPaCa-2, and BxPC-3 ) – A Review
 , Kaniga Pandi1*, Kunal Rupesh Kumar Kataria1, Yokesh Shanmugam2, Maha Swetha Kannuchamy2, Mahalakshmi Sundarraj1, Kaviya Jothi Arumugasamy1, Kaviyapriya Gopal Kuberaselvam1, Praveen Kumar Ramesh1, Bhuvanesh Raj1
, Kaniga Pandi1*, Kunal Rupesh Kumar Kataria1, Yokesh Shanmugam2, Maha Swetha Kannuchamy2, Mahalakshmi Sundarraj1, Kaviya Jothi Arumugasamy1, Kaviyapriya Gopal Kuberaselvam1, Praveen Kumar Ramesh1, Bhuvanesh Raj1 1Department of Pharmaceutical Chemistry, Saveetha College of Pharmacy, Saveetha Institute of Medical and Technical Sciences, Chennai, Tamil Nadu, India.
2Department of Pharmacology, Saveetha College of Pharmacy, Saveetha Institute of Medical and Technical Sciences, Chennai, Tamil Nadu, India.
Corresponding Author E-mail:kanigapharma@gmail.com
DOI : http://dx.doi.org/10.13005/bpj/3420
Download this article as:
![]()
Pancreatic cancer is one of the most lethal malignancies, accounting for approximately 3–4% of all cancers but nearly 7–8% of cancer-related deaths worldwide, with a 5-year survival rate of less than 12%. Its aggressive biology, early metastatic spread, and profound resistance to chemotherapy contribute to poor clinical outcomes. Due to these challenges, in vitro pancreatic cancer cell line models have become indispensable tools for elucidating molecular mechanisms underlying tumor progression and drug resistance. This review comprehensively evaluates three widely used pancreatic cancer cell lines PANC-1, MiaPaCa-2, and BxPC-3 highlighting their distinct molecular, phenotypic, and genetic characteristics and their relevance in anticancer drug discovery. PANC-1 cells, characterized by KRAS mutations and high mesenchymal marker expression, are extensively utilized to study epithelial–mesenchymal transition (EMT), migration, and invasiveness. MiaPaCa-2 cells exhibit enhanced glycolytic activity, stemness-associated markers, and pronounced chemoresistance, making them valuable for investigating cancer stem cell behavior and metabolic adaptations. In contrast, BxPC-3 cells, which are KRAS wild-type, serve as an important model for assessing drug sensitivity and therapeutic response mechanisms, particularly to gemcitabine and targeted agents. Comparative analyses across these models have significantly advanced understanding of key drug resistance pathways, including EMT-driven resistance, altered metabolic signaling, and survival pathway activation, and have facilitated preclinical screening of standard chemotherapeutics and emerging novel compounds. Furthermore, cross-model comparisons provide insights into tumor heterogeneity and support the identification of predictive biomarkers for therapeutic outcomes. Although conventional cell lines remain essential molecular tools, their inability to fully recapitulate the tumor microenvironment limits translational relevance. Therefore, integrating advanced systems such as organoids, patient-derived xenografts, and co-culture models is crucial to bridge the gap between preclinical research and clinical application and to improve therapeutic strategies for pancreatic cancer.
KEYWORDS:Anticancer drug screening; BxPC-3; Drug resistance; MiaPaCa-2; Pancreatic cancer; PANC-1; Targeted therapy
Introduction
Pancreatic cancer is the 12th most common cancer worldwide and the seventh leading cause of cancer mortality.¹ Its poor prognosis is largely due to late-stage diagnosis, with a five-year survival rate below 10%, rapid metastasis, and resistance to conventional therapies. With an estimated 9.1 million disability-adjusted life years in 2017,² it represents a major global disease burden.³The low treatment success rate is attributed to its aggressive clinical course, poor response to chemotherapy or radiotherapy, deep anatomical location, limited tissue accessibility, and absence of reliable screening methods.³ The lifetime cumulative risk is about 1 in 64.⁴ Modifiable risk factors include chronic pancreatitis, obesity, diabetes, smoking, and chemical exposures, while age, gender, family history, and genetic disorders are non-modifiable.⁵ Early-onset pancreatic cancer is increasingly reported among younger individuals,⁶ often associated with aggressive histology and poor prognosis.⁷Surgery with adjuvant chemotherapy remains the standard for resectable pancreatic ductal adenocarcinoma (PDAC).⁸ However, most patients are diagnosed with advanced disease, and only 15–20% undergo surgery; even then, recurrence within a year is common.⁹ Gemcitabine, long considered the gold standard, improved survival compared with 5-fluorouracil (5-FU).¹⁰ More recently, gemcitabine plus nab-paclitaxel and the FOLFIRINOX regimen (5-FU, leucovorin, irinotecan, oxaliplatin)¹¹,¹² have become standard for metastatic disease. A modified FOLFIRINOX protocol as adjuvant therapy showed superior outcomes, with higher disease-free survival (21.6 vs. 12.8 months) and overall survival (54.4 vs. 35.0 months) compared with gemcitabine.¹³Effective clinical care is still significantly hampered by inherent chemoresistance and the asymptomatic nature of early-stage illness, despite advancements in medical imaging and treatment protocols.14 In translational cancer research, cell lines are essential because they act as a link between fundamental laboratory results and practical uses.A controlled and repeatable platform for researching the cellular and molecular mechanisms of cancer genesis, progression, and response to treatment is provided by these in vitro models.Cell lines allow researchers to analyze the intricate signaling pathways that contribute to tumor growth, invasion, and medication resistance in the setting of pancreatic cancer.15They also facilitate high-throughput screening of novel compounds, genetic manipulations using CRISPR or RNA interference, and evaluation of combination therapies before progressing to in vivo studies. Moreover, pancreatic cancer cell lines reflect distinct genetic and phenotypic features of the disease, allowing for subtype-specific investigations. As a result, they are indispensable tools in identifying potential therapeutic targets, validating biomarkers, and accelerating the translation of benchside discoveries into effective clinical strategies.This review’s objective is to examine the biological and molecular traits of these widely used pancreatic cancer cell lines, emphasising their value for researching tumor growth and treatment resistance.By doing so, we underscore their utility in preclinical drug screening, biomarker discovery, and development of targeted therapies aimed at improving patient outcomes.
Materials And Methods
A literature search was performed in PubMed, Scopus, Web of Science, and Google Scholar for studies published between January 2000 and July 2025 on the molecular features, aggressiveness, and chemoresistance of pancreatic cancer cell lines. Keywords included “PANC-1,” “MiaPaCa-2,” “BxPC-3,” “pancreatic cancer cell lines,” “tumor aggressiveness,” “drug resistance,” “KRAS/MAPK,” “PI3K/AKT/mTOR,” “hypoxia,” “HIF-1α,” “VEGF,” “DNA damage repair,” “ATM/ATR,” and “BRCA.”Only peer-reviewed articles in English were included.To find further pertinent studies, the reference lists of the publications that were obtained were also examined.Studies focusing on molecular pathways, signaling differences, and resistance mechanisms in these cell lines were prioritized, while non-relevant, duplicate, or non-original research reports were excluded.
An Overview of Cell Lines for Pancreatic Cancer
PANC-1
The human pancreatic cancer cell line PANC-1 has been the subject of much research.It is produced from a primary pancreatic ductal adenocarcinoma(Fig.1). It harbors a KRAS mutation, a hallmark of pancreatic cancer, which drives oncogenic signaling and contributes to its aggressive behavior. PANC-1 cells exhibit a predominantly mesenchymal phenotype, characterized by reduced cell–cell adhesion and enhanced migratory capacity, making them a valuable model for investigating epithelial-to-mesenchymal transition (EMT). The pancreatic ductal adenocarcinoma cell lines’ genomic and phenotypic characteristics are used to assess how well they respond to peptide receptor radionuclide therapy and neuroendocrine chemotherapy. MIA PaCa 2, which is polymorphic, expresses the mesenchymal marker vimentin and the epithelial markers CK5.6, AE1/AE3, and E cadherin, according to immunohistochemistry and flow cytometry. Additionally, it exhibits neuroendocrine differentiation by expressing synaptophysin and chromogranin-A, as well as hormonal receptors (SSTR2 and NTR1), yet it is devoid of CD56. On the other hand, PANC 1, which exhibits pleomorphism, expresses vimentin, chromogranin-A, CD56, MNF 116, CK5.6, & SSTR2, but not NTR1, E cadherin, or synaptophysin. Both lines feature significant genetic alterations typical of pancreatic cancer, such as homozygous deletion of exons 1-3 of CDKN2A/p16INK4A and KRAS and TP53 mutations, despite the fact that neither line shows microsatellite instability or SMAD4/DPC4 mutations.Importantly, both cell lines express SSTR2 receptors and show neuroendocrine development, which supports their utility as in vitro models for investigating targeted treatments based on somatostatin in pancreatic cancer.16Morphological and biochemical characterization of the PANC‑1 cell line to determine whether it retains key features of differentiated human pancreatic ductal epithelium. The authors found that PANC‑1 cells maintain epithelial-class intermediate filaments, exhibit basolateral localization of Na⁺/K⁺‑ATPase, form complete tight junctions, and adopt a cuboidal morphology when cultured on collagen-coated supports or basement membrane substrates, mirroring ducts observed in situ.
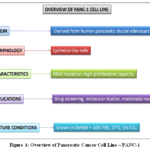 |
Figure 1: Overview of Pancreatic Cancer Cell Line – PANC-1 |
Biochemically, PANC‑1 cells express γ‑glutamyl transpeptidase, carbonic anhydrase, and functional Na⁺/K⁺‑ATPase activity assessed via [³H]ouabain binding resembling enzymatic profiles of native rat and human pancreatic ducts. Additionally, they synthesize and secrete high‑molecular‑weight sulfated proteins (ranging ~180 kDa to 1 MDa), with a predominant species of ~660 kDa, demonstrating secretory behavior consistent with ductal epithelium.Comparative analyses revealed that PANC‑1 cells closely resemble other ductal-origin lines (e.g., MDCK), and differ markedly from mesenchymal-derived lines such as L cells, confirming their epithelial lineage. The findings suggest that PANC‑1 retains several fundamental differentiated traitsstructural, enzymatic, and secretory making it a valuable model for investigating ductal secretion and epithelial function in vitro.17PANC‑1 derivatives stably expressing either a dominant‑negative HRAS mutant (HRAS^S17N), a constitutively active KRAS^G12V, or an EGFP control, then treated these cells with TGF‑β1 and profiled gene expression changes across three independent experiments. Their findings revealed that activation state of RAS critically influenced TGF‑β1–driven responses: Genes related to extracellular matrix remodelling, cell adhesion, and epithelial–mesenchymal transition (EMT) showed increased transcriptional activation in cells with active KRAS^G12V when compared to controls; dominant negative HRAS mitigated these effects.In functional terms, this meant that constitutive KRAS activation potentiated TGF‑β1‑induced EMT-like features including upregulation of mesenchymal markers and motility‑related genes while HRAS inhibition reduced these phenotypic shifts. These results emphasize a cooperative interplay between oncogenic RAS signaling and TGF‑β1 pathways in driving EMT and aggressive behavior in PANC‑1 cells, providing mechanistic insight into how KRAS mutations promote tumor progression in pancreatic ductal adenocarcinoma.18Along with EMT-type cells, a subpopulation of pancreatic cancer stem cells (CSCs) identified by markers like CD44, CD24, ESA, and CD133 are crucial for the development of metastatic pancreatic cancer and therapeutic resistance.CSCs possess defining features of self-renewal, multipotency, and high tumorigenicity, and are inherently more resilient to conventional chemoradiotherapy. EMT is typified by an overexpression of mesenchymal factors, which also encourage invasiveness, and a downregulation of epithelial markers such E cadherin(like vimentin, Zeb 1/Slug) also seems to give tumor cells stem-like characteristics, which supports a CSC phenotype.This dual CSC/EMT phenotype contributes significantly to both intrinsic and acquired chemoresistance as well as metastatic potential. Consequently, the authors argue that targeting CSCs and reversing EMT phenotypes are promising strategies to enhance drug sensitivity, suppress metastasis, and ultimately improve clinical outcomes in pancreatic ductal adenocarcinoma (PDAC).19 Due to their plasticity and expression of stemness-associated markers, PANC-1 cells are frequently used in studies exploring cancer stem cell properties and tumor heterogeneity. Furthermore, they display intrinsic resistance to chemotherapeutic agents like gemcitabine, positioning them as a robust model for evaluating mechanisms of drug resistance and testing novel therapeutic strategies aimed at overcoming treatment failure in pancreatic cancer.
MiaPaCa-2
MiaPaCa-2 is a human pancreatic ductal adenocarcinoma cell line widely utilized in cancer biology due to its well-characterized genetic and metabolic profile (Fig.2). Originating from a moderately differentiated pancreatic carcinoma, MiaPaCa-2 cells harbor a KRAS mutation, which drives oncogenic signaling and contributes to their aggressive phenotype. One of the defining features of this cell line is its high glycolytic activity, making it an ideal model for studying the metabolic reprogramming commonly seen in pancreatic tumors. MiaPaCa-2 cells also exhibit significant mitochondrial dysfunction and oxidative stress, which has made them a valuable tool in research focusing on cancer cell metabolism, mitochondrial targeting, and apoptosis.
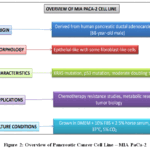 |
Figure 2: Overview of Pancreatic Cancer Cell Line – MIA PaCa-2 |
The functional influence on PDAC progression and the clinical importance of fibroblast activation protein-α (FAP) stromal expression. 98% of 48 PDAC specimens had stromal FAP expression, according to immunohistochemical examination; the expression varied in intensity from weak to strong and was not present in non-cancerous pancreatic tissue. Overall survival was considerably lower for patients with moderate to strong FAP expression (mean survival ≈352 days) than for those with weak or negative expression (≈497 days; p = 0.006). The scientists created human FAP-expressing NIH 3T3 fibroblast lines and cocultured them indirectly that is, without direct cell contact with MiaPaCa 2 pancreatic cancer cells in order to investigate possible causes.As opposed to coculturing with fibroblasts that were control and did not express FAP, coculture with fibroblasts that expressed FAP markedly increased the invasiveness of MiaPaCa 2.Additionally, by encouraging the phosphorylation of the retinoblastoma (Rb) protein, FAP-expressing fibroblasts sped up the cell cycle progression of MiaPaCa 2 cells, causing them to transition from G₀/G₁ to S/G₂/M phases.Together, these findings imply that stromal FAP expression is a negative prognostic indicator in PDAC and functionally contributes to tumor aggressiveness by enhancing cancer cell invasion and proliferation through paracrine stromal–epithelial interactions.20In MiaPaCa 2 human pancreatic adenocarcinoma cells, the anticancer activity and underlying molecular mechanisms of Tan IIA, a bioactive chemical produced from Salvia miltiorrhiza. Tan IIA treatment markedly reduced the expression of EGFR, IGF1R, VEGFR, Ras, Raf, MEK, ERK, PI3K, AKT, and mTOR, while also affecting PTEN. Molecular analyses revealed that Tan IIA inhibits the Ras/Raf/MEK/ERK and PI3K/AKT/mTOR pathways, key drivers of pancreatic cancer survival and proliferation.As a result, MiaPaCa‑2 cells exposed to Tan‑IIA exhibited marked reductions in growth and viability, indicating that targeting these parallel kinase cascades may be an effective strategy for suppressing tumor aggressiveness in this model.21A novel strategy to impair oncogenic KRAS signaling in pancreatic cancer by targeting lipid metabolism, specifically focusing on phosphatidylserine (PS) remodeling. They show that the enzyme LPCAT3, responsible for generating unsaturated PS species essential for KRAS nanocluster formation at the plasma membrane, is a key dependency in KRAS‑mutant pancreatic cancer cells such as MiaPaCa‑2. Silencing LPCAT3 via siRNA or CRISPR, as well as using small-molecule inhibitors (HTS3/HTS4), led to depletion of unsaturated PS, mislocalization of mutant KRAS from the membrane, disruption of KRAS nanoclustering, and suppression of MAPK signaling. This resulted in significantly reduced proliferation and colony-forming ability in KRAS-dependent tumor cells, with minimal effects in KRAS wild-type cells. These findings highlight LPCAT3 inhibition as a potential pan-KRAS-targeting therapeutic approach that bypasses allele-specific resistance mechanisms and disrupts KRAS-driven tumorigenesis by perturbing essential lipid-mediated nanoscale architecture on the plasma membrane.22Through a variety of ways, it has been demonstrated that the naturally occurring anthraquinone compound aloe emodin has strong anticancer effects on the human pancreatic adenocarcinoma cell lines. Cell viability and colony formation were significantly reduced as a result of the treatment, with MiaPaCa-2 cells showing higher sensitivity than PANC-1. Aloe emodin caused chromatin condensation, membrane blebbing, and cell shrinkage all of which are obvious morphological changes associated with apoptosis.Both early and late apoptotic cell populations significantly increased, according to flow cytometry data, and there was a noticeable buildup of cells in the sub-G1 phase, which suggests apoptotic DNA fragmentation. Importantly, aloe emodin disrupted the mitochondrial membrane potential (ΔΨm), indicating activation of the intrinsic apoptotic mechanism. Aloe emodin increased autophagy and promoted apoptosis, as evidenced by increased LC3-II expression.The dual activation of apoptotic and autophagic pathways, combined with cell cycle arrest and mitochondrial dysfunction, highlights aloe emodin’s multi-targeted anticancer potential, especially against KRAS-mutated pancreatic cancer cells.23Because of their sensitivity to metabolic inhibitors and pro-apoptotic stimuli, MiaPaCa-2 cells are frequently employed in preclinical studies aimed at developing therapies that exploit cancer-specific metabolic vulnerabilities.
BxPC-3
BxPC-3 was established from a moderately differentiated adenocarcinoma (Fig.3). It stands out from many previous pancreatic cancer models because it has KRAS wild-type. In contrast to more mesenchymal or stem-like cell lines like PANC-1 or MiaPaCa-2, it has a well-defined epithelial phenotype, tight cell–cell junctions, and significant E-cadherin expression.BxPC-3 is an important model for studying KRAS-independent signalling pathways and assessing the effectiveness of targeted treatments that might not depend on the RAS/MAPK axis because it lacks KRAS mutations. BxPC-3 is widely used by researchers to investigate receptor tyrosine kinase signalling (EGFR, HER2), alternative oncogenic drivers, and the cellular response to chemotherapeutics in situations when KRAS is not present. Exposure of human CD14+ monocyte derived dendritic cells (DCs) to conditioned media from the highly metastatic pancreatic cancer cell line BxPC 3 (BxCM) resulted in impaired DC differentiation, maturation, and antigen-presentation capacity and increased microRNA 146a (miR 146a).Specifically, DCs treated with BxCM failed to fully differentiate or effectively stimulate antigen-specific T‑cell responses key functions required for effective immunity. Importantly, inhibition of miR‑146a expression partially reversed these functional impairments, suggesting that elevated miR‑146a plays a central role in mediating BxCM-induced DC dysfunction, likely through downregulation of the SMAD4 signaling pathway. These results identify miR‑146a as a critical suppressive factor induced by KRAS‑independent stromal signaling, highlighting how BxPC‑3 tumors can orchestrate immunosuppressive microenvironments through miRNA-mediated reprogramming of DCs.24With an IC₅₀ of approximately 19.3 µg/mL and up to ~90 % inhibition at 32 µg/Ml, oridonin, a diterpenoid derived from Rabdosiarubescens, effectively inhibited cellular growth in a dose-dependent manner, according to MTT studies.In addition to its cytotoxic effects, oridonin greatly decreased the secretion of pro-inflammatory cytokines, including IL 1β, IL 6, and IL 33, dose-dependent reductions were verified by ELISA (e.g., IL 1β declined from approximately 14.40 to approximately 11.17 pg/mL; IL 6 decreased from approximately 4.45 to approximately 3.95; IL 33 decreased similarly).At the molecular level, the therapy increased the expression of Smad4 while suppressing TGF β1, BMP 2, and several Smad proteins. Important nuclear transcription factors and inflammatory mediators, including AP 1, RelA (and phosphorylated RelA), NF-κB1, STAT3, and phosphorylated STAT3, were similarly downregulated.Furthermore, after receiving oridonin, the expression of tumor-associated hallmark proteins such MMP 2, VEGF, and survivin was reduced.Taken together, these findings indicate that oridonin exerts anticancer effects in BxPC‑3 cells through dual mechanismsinhibition of inflammation-associated cytokines and disruption of transcription factor networkseffectively targeting cancer hallmarks like growth, angiogenesis, invasion, and metastasis in a KRAS‑wild‑type epithelial pancreatic cancer context.25The role of Response Gene to Complement 32 (RGC 32) in TGF-induced epithelial mesenchymal transition (EMT) and the migration of the human pancreatic cancer cell line BxPC 3. Their immunohistochemical examination of tumor samples showed that in patients with pancreatic cancer, greater TNM stage and lymph node metastases were substantially connected with abnormal E cadherin localisation and increased RGC 32 expression. Exposure to TGF β₁ caused a dose- and time-dependent rise in RGC 32 in cultivated BxPC 3 cells, along with the EMT hallmarks of vimentin overexpression and E cadherin downregulation. Significantly, in transwell tests, siRNA-mediated reduction of RGC 32 decreased migratory capacity and significantly hampered TGF β-induced EMT alterations.However, since BxPC 3 cells lack functioning Smad4, overexpression of RGC 32 alone was enough to induce EMT marker changes and improve cell migration even in the absence of exogenous TGF β₁. This suggests that RGC 32 can initiate EMT via a KRAS independent, Smad independent mechanism.According to these findings, RGC 32 is a new gene that promotes metastases in pancreatic cancer by boosting TGF-β-induced EMT and making BxPC 3 cells of the KRAS wild type more aggressive.26No matter if a KRAS mutation is present or not, in vivo serial passage of human pancreatic cancer cell lines in orthotopic animal models could enrich aggressive metastatic phenotypes. They extracted tumour from critically ill mice, serially re-implanted derived cells for up to six passages, and orthotopically implanted 10^6 BxPC 3 RFP (KRAS wild-type) or PANC 1 GFP (KRAS-mutant) cells into the pancreas tail of naked animals.By the sixth passage (P6 for BxPC‑3, P5 for PANC‑1), both cell lines gave rise to significantly more aggressive tumours, characterized by accelerated primary growth, dramatically shortened median survival (from ~127 days to ~52 days in BxPC‑3; from ~124 days to ~42 days in PANC‑1), and metastasis detectable as early as three weeks post-implantation (versus ~14 weeks in parental lines). This study compellingly demonstrates that serial in vivo selection yields highly metastatic variants irrespective of KRAS mutation, making these derivatives powerful models to study the molecular basis of aggressive pancreatic cancer and to rigorously test potential therapeutic interventions.27The KRAS signalling system is modulated in pancreatic ductal adenocarcinoma models, including KRAS mutant (MiaPaCa 2, PaCa 44) and KRAS wild type (BxPC 3) cell lines, as well as in vivo xenograft models, by treatment with both standard paclitaxel and the investigational taxane SB T 1216.
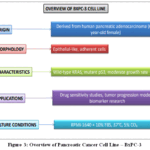 |
Figure 3; Overview of Pancreatic Cancer Cell Line – BxPC-3 |
According to their findings, the baseline expression of genes linked to KRAS downstream branches, including PI3K/PDK1/AKT, RalGEF, and RIN1/ABL, differed significantly between cell lines based on the presence or absence of KRAS mutations. Nevertheless, taxanes consistently led to downregulation of the KRAS signalling pathway at the mRNA and protein levels, even in the absence of a KRAS mutation.There was no correlation between KRAS mutational status and differential sensitivity to taxane treatment, suggesting that taxanes act through mechanisms beyond KRAS-dependent pathways. Importantly, the study concluded that despite effective suppression of KRAS-related pathway expression, no predictive biomarkers for taxane activity or targets to sensitize PDAC models were identified via KRAS pathway profiling.28A novel PET imaging strategy using zirconium-89-labeled transferrin ([⁸⁹Zr]Zr‑Tf) to noninvasively assess oncogenic signaling and target engagement in PDAC. The study used the MYC-induced expression of the transferrin receptor (TfR) as a quantifiable biomarker because KRAS mutations are seen in more than 90% of PDAC cases and drive downstream pathways including ERK and MYC.In both genetically engineered mouse models and human PDAC xenografts, high uptake of [⁸⁹Zr]Zr‑Tf was observed in tumours with active KRAS signaling. This uptake significantly decreased upon treatment with JQ1 (a BRD4 inhibitor) or SCH772984 (an ERK inhibitor), indicating reduced MYC and TfR expression. Notably, KRAS-mutant xenografts such as Capan-2 and Suit-2 showed strong tracer signals and sensitivity to inhibition, while KRAS wild-type models like BxPC-3 exhibited low-to-moderate uptake that was unaffected by these inhibitors. Immunohistochemical analyses confirmed that PET tracer accumulation correlated with TfR and MYC protein levels in the tumours. These findings demonstrate that [⁸⁹Zr]Zr‑Tf PET imaging can serve as a functional tool for evaluating KRAS–MYC pathway activity and therapeutic responses, offering a promising approach for precision oncology in pancreatic cancer.29This cell line is thus particularly valuable for developing and testing therapeutic strategies that may benefit the subset of pancreatic cancer patients lacking KRAS mutations.
Molecular Pathways Involved In Aggressiveness
EMT markers
When epithelial cells lose their polarity and cell-cell adhesion properties and develop mesenchymal traits that improve their migratory and invasive capacities two crucial elements of cancer growth and metastasis a fundamental biological process known as the epithelial–mesenchymal transition (EMT) takes place (Fig.4). This transition is indicated by a noticeable alteration in molecular markers. The development of embryos and the maintenance of proper tissue architecture depend on calcium-dependent cell adhesion molecules known as cadherins. Additionally, they support the preservation of various tissues’ epithelial structure.30Since some carcinomas have been found to have an aggressive and undifferentiated character due to lack of E-cadherin expression, including pancreatic carcinoma, it is well known that E-cadherin contributes to tumour progression and metastasis.31Another adhesion molecule, N-cadherin (neural cadherin), is linked to an increased propensity for invasion in cancer.E-cadherin, a hallmark epithelial marker responsible for maintaining intercellular adhesion, is typically downregulated during EMT, contributing to the loss of epithelial integrity. Conversely, N-cadherin, a mesenchymal cadherin, is upregulated, promoting increased cell motility and invasiveness, a phenomenon often referred to as the “cadherin switch.” Vimentin, an intermediate filament protein, is another key mesenchymal marker that becomes highly expressed in cells undergoing EMT, further supporting changes in cell shape, structural flexibility, and metastatic potential. Dynamic cell mobility and tissue patterning depend on adhesion changes regulated by growth factors. E-cadherin expression is modulated by such signals; for example, TGF-β induces epithelial-to-fibroblastic transition with reduced E-cadherin,³²while FGF-1 and FGF-2 enhance E-cadherin-mediated adhesion and reduce cancer cell invasion.³³Furthermore, FGF receptor stimulation may promote N-cadherin-dependent motility, although the mechanism governing cadherin expression is unknown.34The large number of nerves inside and surrounding the organ is most likely one of the primary causes of pancreatic cancer spreading along the neural bands.The adhesion molecules that determine the cancer cells’ affinity for the neural band may also have an impact on the cancer cells’ motility. One study found no connection between neural invasion and the expression of neural cell adhesion molecules.35N-cadherin, normally abundant in neuronal and mesenchymal tissues, is closely linked to pancreatic cancer invasiveness and neural invasion.36In aggressive, drug-resistant cell lines like PANC-1 and MiaPaCa-2, reduced E-cadherin with elevated N-cadherin and Vimentin marks EMT activation.
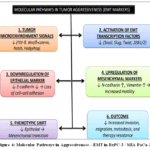 |
Figure 4: Molecular Pathways in Aggressiveness – EMT in BxPC-3 / MIA PaCa-2 |
Stemness (SOX2, OCT4, NANOG)
The ability of stem cells (SCs) to self-renew and generate progeny that can follow a variety of differentiation paths is one of their traditional definitions.37There are now four types of SCs recognised.38The first two, known as somatic and embryonic stem cells , are biologically present at various periods of life. Since ESCs are the most researched SCs, the information gleaned from their study has influenced research on other SC types.ASCs, postnatal derivatives of ESCs, retain expression of key transcription factors OCT4, KLF4, and SOX2. In pancreatic cancer, stemness is defined by the expression of OCT4, SOX2, and NANOG, which maintain CSC self-renewal and pluripotency, contributing to tumourigenicity, metastasis, and therapy resistance. These markers are elevated in aggressive PDAC lines like PANC-1 and MiaPaCa-2.Recent studies highlight nine transcription factors essential for maintaining pluripotency in mouse cells—OCT4, NANOG, SOX2, Dppa4, Dppa5, Sall4, Utf1, Rex2, and Rif1.39OCT4, NANOG, and SOX2 are highly expressed in the ICM during early embryogenesis, sustaining ESC pluripotency.40All stem cell types share these core pluripotency genes, while c-MYC, a key oncogene, exhibits greater variability but regulates many stem cell properties.41These genes control transcription factors, surface markers, ABC transporters, and enzymes that define stem cell behavior.42Although CSC regulation is well studied across cancers, few publications address SOX2, OCT4, and NANOG in pancreatic cancer, with 24, 27, and 20 studies found in PubMed, respectively. Elevated OCT4 and NANOG correlate with early carcinogenesis and poor prognosis,43,44while OCT4 also mediates metastasis and multidrug resistance. Cells positive for NANOG, OCT4, and SOX2 show enhanced growth, invasion, and drug resistance.45,46OCT4 helps to maintain pluripotency and supports the long-term survival of CSCs, whereas SOX2 is essential for maintaining the undifferentiated state and accelerating tumour growth. NANOG supports the transcriptional network that maintains the stem-like phenotype by acting downstream of SOX2 and OCT4.The coordinated overexpression of these stemness markers in pancreatic cancer cells not only drives tumour heterogeneity and relapse but also presents promising targets for therapeutic intervention aimed at eliminating CSC populations and improving patient outcomes.
 |
Figure 5; SOX2, NANOG, and OCT4 show overlapping expression across all four stem cell types :embryonic stem cells (ESCs), adult stem cells (ASCs), induced pluripotent stem cells (iPSCs), and cancer stem cells (CSCs) |
Invasion-related genes (MMPs, CXCR4)
Invasive genes, like matrix metalloproteinases (MMPs) and The promotion of pancreatic cancer cell invasion and metastasis depends on C-X-C chemokine receptor type 4.MMPs, especially MMP-2 and MMP-9, are enzymes that break down extracellular matrix components, which helps tumourcells invade neighbouring tissues and spread (Fig.6). Their overexpression in pancreatic ductal adenocarcinoma (PDAC) is often linked to poor prognosis, lymph node involvement, and advanced tumour stage.Similarly, in response to its ligand CXCL12 (SDF-1), the chemokine receptor CXCR4 promotes directed cell migration. In PDAC cell lines, high CXCR4 expression facilitates chemotactic migration towards distant organs and encourages the formation of metastatic lesions. In pancreatic ductal adenocarcinoma (PDAC), the CXCL12 (SDF 1α)/CXCR4 chemotactic axis is essential for promoting perineural invasion (PNI).
 |
Figure 6: Molecular Pathways in Pancreatic Cancer Aggressiveness: Invasion-related Genes |
Analysis of 78 human PDAC specimens showed that elevated CXCR4 expression strongly correlated with PNI occurrence, suggesting its clinical relevance. generated by peripheral nerves CXCL12 exhibited paracrine activity in both in vitro and in vivo models, facilitating the migration and invasion of cancer cells that were positive for CXCR4 and encouraging the formation of dorsal root ganglia in the direction of tumourcells.Importantly, pharmacologic or genetic inhibition of CXCR4 signaling significantly suppressed tumour invasion and halted sciatic nerve extension in orthotopic mouse models, underscoring its therapeutic potential for preventing PNI. These findings position the CXCL12‑CXCR4 pathway as a critical mediator of nerve tropism and invasion in pancreatic cancer, highlighting it as a promising target to curtail local neuroinvasion and disease progression.47In vitro, pancreatic cancer cell lines, including KRAS wild type BxPC 3 and KRAS-mutant MiaPaCa 2/PANC 1, showed markedly reduced proliferation, colony formation, and invasion when the stromal derived factor 1α (SDF 1/CXCL12)–CXCR4 axis was blocked by shRNA or pharmacological inhibition. Importantly, CXCR4 inhibition resulted in a downregulation of canonical Wnt/β catenin signalling, as seen by decreased expression of mesenchymal markers including vimentin and Slug, as well as decreased TOPflash reporter activity and Wnt target gene expression. These findings imply that because CXCR4 regulates the Wnt/β catenin pathway, which in turn encourages EMT and invasive behaviour, it may be a suitable therapeutic target for halting the progression of pancreatic cancer.48According to recent studies, CXCR7 (also called ACKR3) and CXCR4 often co-express in pancreatic cancer tissues and cell lines. They also work together to transmit CXCL12-driven signalling that improves the behaviour of tumourcells.Upon binding CXCL12 or CXCL11, CXCR7 can activate MAPK/ERK pathways via β-arrestin–dependent mechanisms, while CXCR4 primarily transduces Gαi-mediated signals that activate KRAS and MAPK/ERK; both receptors together amplify downstream proliferative and migratory outputs. Functionally, this co‑expression appears to promote EMT and directional metastasis, with CXCR7 enhancing CXCR4 signaling fidelity by shaping the CXCL12 gradient through ligand sequestration thereby influencing cell motility and invasion along defined pathways. The mechanistic insights strongly support the interpretation that CXCR7 assists CXCR4 in modulating TGF‑β‑induced EMT, mesenchymal transition, and tissue‑directed metastatic spread in pancreatic cancer models expressing both receptors.49The elevated expression of MMPs and CXCR4 is primarily responsible for the aggressive nature of pancreatic cancer, making them essential molecular targets for anti-metastatic therapies (Fig.7).
 |
Figure 7: Molecular Pathways Driving Tumour Invasion via MMPs and CXCR4 |
Hypoxia pathways (HIF-1α, VEGF)
Solid tumours, such as pancreatic cancer, are characterised by hypoxia, which greatly increases the tumour’s aggressiveness, resistance to treatment, and potential for metastasis (Fig.8). Low oxygen levels cause the hypoxia-inducible factor 1-alpha (HIF-1α) to stabilise and migrate to the nucleus, where it initiates the transcription of several target genes involved in hypoxia adaptation. One of its primary downstream targets is vascular endothelial growth factor (VEGF), a potent angiogenic agent that promotes the formation of new blood vessels to support the oxygen-depleted tumour.In pancreatic cancer cell lines, upregulation of HIF-1α and VEGF has been connected to increased angiogenesis, epithelial–mesenchymal transition (EMT), and stemness, all of which have been connected to invasion and metastasis.To further improve tumour survival and progression, the hypoxic microenvironment also interacts with additional oncogenic signaling pathways, including RAS/RAF/MEK/ERK and PI3K/AKT/mTOR.Hypoxia resulting from rapid tumour growth and dense desmoplastic stroma is a key driver of angiogenesis in pancreatic ductal adenocarcinoma (PDAC). Stabilization of HIF‑1α under low‑oxygen conditions triggers transcriptional upregulation of VEGF and other pro‑angiogenic genes, fostering the formation of an abnormal, leaky vascular network that paradoxically fails to relieve hypoxia but promotes tumour aggressiveness. Elevated HIF‑1α expression in PDAC has been correlated with regions of fibrosis and high microvessel density, further associating hypoxia with enhanced local invasion, metastatic spread, and poor prognosis. While early anti‑angiogenic strategies targeting VEGF or its receptors (e.g. with tyrosine kinase inhibitors or VEGFR antibodies) demonstrated preclinical efficacy, clinical trials have yielded disappointing results when used alone highlighting the need for combination regimens (e.g. VEGF blockade plus MMP inhibitors or chemotherapy) to effectively disrupt this complex, hypoxia-driven angiogenic network.50Hypoxia-inducible factor 1 (HIF 1), and particularly its α subunit, is an essential mediator that links tumour hypoxia to VEGF production and angiogenesis in human pancreatic ductal adenocarcinoma models and tissue samples.In human pancreatic cancer samples, elevated HIF‑1α protein localized to tumour cell nuclei and strongly correlated with increased VEGF mRNA in the same cancer cells, indicating coordinated activation under hypoxic conditions. Several human pancreatic cancer cell lines were exposed to low oxygen conditions in vitro, which resulted in high HIF 1 DNA-binding activity at the VEGF promoter and increased amounts of VEGF mRNA and protein.Cells with high constitutive HIF‑1α also produced higher baseline VEGF, even under normoxia. These results establish HIF‑1α as the molecular switch that activates VEGF-driven neoangiogenesis, thus connecting intratumoural hypoxia with abnormal vascularization and aggressive tumour behavior in pancreatic cancer.51Therefore, targeting hypoxia-driven pathways particularly HIF-1α and VEGF remains a promising therapeutic approach in overcoming the aggressive nature of pancreatic ductal adenocarcinoma.
 |
Figure 8: Hypoxia Pathways Driving Cancer Aggressiveness |
KRAS/MAPK and PI3K/AKT/mTOR signaling differences between the cell lines
Commonly utilised cell lines have very different signalling characteristics in pancreatic cancer research, especially in the KRAS/MAPK and PI3K/AKT/mTOR pathways (Table 1).
Table 1: Molecular Pathways Involved in Aggressiveness: KRAS/MAPK vs PI3K/AKT/mTOR
| Feature | KRAS/MAPK Pathway | PI3K/AKT/mTOR Pathway |
| Primary Role | Proliferation, differentiation, migration | Survival, metabolism, growth |
| Upstream Trigger | KRAS mutation/EGFR activation | PI3K activation/PTEN loss |
| Key Steps | KRAS → RAF → MEK → ERK → Gene transcription | PI3K → PIP3 → AKT → mTOR → Protein synthesis |
| Effect in Aggressive Cell Lines | ↑ EMT, ↑ invasion potential | ↑ Anti-apoptotic signaling, ↑ metabolic adaptation |
| Observed Difference Between Cell Lines | Cell line A: Higher ERK phosphorylation; Cell line B: Lower ERK | Cell line A: Moderate AKT activity; Cell line B: Strong AKT & mTOR activation |
MiaPaCa-2 and PANC-1 cells both have activating KRAS mutations. These mutations promote carcinogenic characteristics such migration, proliferation, and resistance to apoptosis and result in constitutive activation of the KRAS/MAPK cascade (RAS–RAF–MEK–ERK).These cell lines are also good models for testing dual-targeted treatments that try to block both pathways at once since they show strong PI3K/AKT/mTOR pathway activity.In contrast, BxPC-3 cells are KRAS wild-type and maintain an epithelial phenotype, relying on alternative mechanisms such as receptor tyrosine kinases for MAPK activation. Their PI3K/AKT/mTOR signaling is less constitutively active, positioning BxPC-3 as a valuable model for studying KRAS-independent therapeutic strategies.52These differences highlight the importance of selecting appropriate cell lines based on their distinct molecular characteristics when designing experiments focused on pancreatic cancer signaling and drug resistance.Pharmacologic inhibition of the KRAS–MAPK pathway in pancreatic ductal adenocarcinoma (PDAC) causes HER2 to compensate, which is driven by the phosphatase DUSP6 being broken down by proteasomes.Under MAPK blockade, the loss of DUSP6 destabilizes the negative regulation of HER2, resulting in rapid HER2 phosphorylation and signaling reactivation. Leveraging this understanding, the researchers show that combining ERK pathway inhibitors (e.g., ulixertinib or trametinib) with trastuzumabderuxtecan an anti‑HER2 antibody–drug conjugate elicits sustained and complete tumour regression in multiple patient-derived xenograft models of PDAC, without significant toxicity. Notably, even KRAS inhibitors led to similar HER2 activation, further supporting the combination approach. These results identify a rational therapeutic strategy dual targeting of KRAS/MAPK and HER2 to overcome adaptive resistance and improve treatment efficacy in pancreatic cancer.53These differences highlight the importance of selecting appropriate cell lines based on their distinct molecular characteristics when designing experiments focused on pancreatic cancer signaling and drug resistance.
Chemoresistance Mechanisms In Each Cell Line
Common drugs: Gemcitabine, 5-FU, Oxaliplatin, Paclitaxel
The multiple processes behind the well-known chemoresistance of pancreatic ductal adenocarcinoma (PDAC) to chemotherapy. Different chemoresistance profiles to common treatment drugs like gemcitabine, 5-fluorouracil (5-FU), oxaliplatin, and paclitaxel are shown by pancreatic cancer cell lines.Patients with advanced and metastatic PC respond effectively to gemcitabine and other treatment drugs, although their effectiveness is significantly reduced when chemoresistance to gemcitabine develops. Compared to other chemotherapy drugs, gemcitabine is without a doubt more effective against pancreatic cancer cells. Most studies on chemoresistance in advanced pancreatic cancer focus on gemcitabine, as data on other drugs remain limited. The mechanisms underlying gemcitabine resistance are not fully understood but are linked to transcription factors, signaling pathways, and enzymes involved in nucleoside metabolism.54EMT plays a key role, where upregulation of Snail, Twist, and Zeb1 and loss of E-cadherin enhance invasiveness and reduce gemcitabine uptake by downregulating nucleoside transporters. Additionally, cancer stem cells (CSCs) contribute to resistance through self-renewal and drug efflux capacity.As a result, CSCs are able to withstand standard treatments. Dysregulated non-coding RNAs (including miR-29c, miR-210, and lncRNAs) that promote autophagy, DNA repair, and survival signaling enable these CSCs.Stromal-derived factors like SDF-1α are secreted by the tumour microenvironment, particularly by pancreatic stellate cells. These factors interact with cancer cells’ CXCR4 and trigger downstream signalling pathways (including PI3K/AKT and ERK) that strengthen drug resistance. A key role is also played by hypoxia, which is mediated by HIF-1α activation and supports EMT and stemness features by encouraging angiogenesis and metabolic adaptability. Furthermore, the intracellular activation of gemcitabine is restricted by changes in drug transporter and metabolic enzyme expression, such as reduced expression of human equilibrativenucleoside transporter 1 and deoxycytidine kinase , whereas increased expression of ATP-binding cassette transporters results in enhanced drug efflux, including MDR1.55MiaPaCa-2, which harbors a mutant KRAS gene and displays high glycolytic activity, often resists gemcitabine due to overexpression of RRM1, reduced nucleoside transporter (hENT1) expression, and enhanced activity of survival pathways like PI3K/AKT and NF-κB. It also demonstrates resistance to 5-FU and oxaliplatin via increased drug efflux (ABCC1/MRP1) and DNA repair mechanisms, alongside enhanced autophagy and stemness (SOX2, OCT4) contributing to cell survival under treatment stress. In contrast, BxPC-3, a KRAS wild-type line, shows relatively higher sensitivity to gemcitabine but acquires resistance through EMT activation and upregulation of N-cadherin and Vimentin, leading to a more invasive phenotype.Oxaliplatin resistance, on the other hand, involves increased synthesis of DNA damage response elements and anti-apoptotic proteins (Bcl-2, survivin), paclitaxel resistance in BxPC-3 is associated with β-tubulin isotype switching and activation of MAPK/ERK signalling.56Both lines demonstrate altered hypoxia responses (HIF-1α, VEGF), which further facilitate chemoresistance by promoting angiogenesis and metabolic adaptation. The poor response of PDAC to chemotherapeutic drugs including gemcitabine, 5-FU, oxaliplatin, and paclitaxel is a result of these coupled processes.57Designing successful combination tactics to overcome medication resistance in pancreatic cancer requires an understanding of these pathways.
Overexpression of efflux transporters (e.g., MDR1/P-gp, MRP1)
The upregulation of efflux transporters, such as multidrug resistance-associated protein 1 (MRP1) and P-glycoprotein (P-gp, encoded by the MDR1 gene), mediates chemoresistance in pancreatic cancer.By vigorously pumping a variety of chemotherapeutic medications out of cancer cells, these ATP-binding cassette (ABC) transporters lessen intracellular drug accumulation and its lethal effects.Elevated P-gp and MRP1 expression has been linked to poor response to popular chemotherapeutic drugs, including gemcitabine, 5-fluorouracil, paclitaxel, and oxaliplatin, in pancreatic ductal adenocarcinoma (PDAC).This overexpression is often driven by genetic and epigenetic changes and is further exacerbated by microenvironmental factors such as hypoxia and inflammatory cytokines. Moreover, cancer stem cell populations within pancreatic tumours tend to express high levels of these efflux proteins, contributing to their intrinsic resistance and the likelihood of relapse following treatment. In human pancreatic cancer specimens, the first systematic immunohistochemical investigation of MDR1/P-glycoprotein (P-gp) and MRP-1 expression showed that 93.3% of tumours express MDR1/P-gp, whereas 31% co-express MRP-1 and MDR1, and a small minority (~6.7%) lack both expressions.Most tumours displayed moderate to strong staining intensity and broad distribution within tumour cells. These high expression rates suggest that MDR1/P-gp, and to a lesser extent MRP-1, are frequently present in pancreatic cancer and may significantly contribute to clinical chemoresistance though a direct causal link remains to be confirmed. The study underscores the importance of considering efflux pump expression when designing chemotherapy regimens or evaluating inhibitors targeting drug resistance mechanisms in pancreatic cancer.58All things considered, focussing on these efflux transporters offers a viable way to get past medication resistance and enhance treatment results for individuals with pancreatic cancer.
Role of autophagy and apoptosis evasion
Pancreatic cancer cells rely on autophagy and apoptosis evasion for survival, growth, and resistance to treatment (Fig.9).Autophagy, a cellular degradation process, helps cancer cells withstand metabolic stress and nutrient deprivation by recycling damaged organelles and proteins. In pancreatic ductal adenocarcinoma (PDAC), autophagy is often upregulated and acts as a survival mechanism, enabling tumour cells to adapt to the hypoxic and nutrient-poor tumour microenvironment. By halting drug-induced cell death, this pathway not only encourages tumour growth but also builds resistance to chemotherapeutic drugs like gemcitabine. PDAC is a complex aetiology that includes gene mutation, chemical exposure, and inflammatory induction. These mechanisms mediate pancreatic carcinogenesis, and a malfunction in autophagy can either promote or hinder it.59Inflammation is one of the most significant extrinsic variables in the development of pancreatic tumours. Autophagy controls inflammation in the early stages of pancreatic cancer in a complex way. Acinar-ductal metaplasia (ADM), a pathogenic change that arises when oncogenic signalling is triggered (e.g., gene mutation and inflammation), is the precursor to PanIN. In cells of the pancreatic epithelium (Pdx1-Cre;atg7fl/fl),conditional deletion of Atg7 causes severe acinar cell degeneration.60This is followed by the development of ADM due to mitochondrial malfunction, ER stress, and decreased protein synthesis.60Pancreatic cancer may emerge as a result of ATG7-mediated inflammatory regulation. By controlling cell death signals, pancreatic Atg7 deletion (Pdx1-Cre;atg7fl/fl) also causes inflammation and fibrosis in mice. Autophagy increases TBK1 (TANK binding kinase 1)-mediated dysplasia, which aids in the carcinogenesis of KRAS-driven pancreatic cancer.62,63Enhanced autophagy reduces TBK1-mediated inflammation in mice, so autophagy inhibition or Atg5 deletion (Pdx1-Cre;atg5^fl/fl) exacerbates pancreatitis and dysplasia.63Thus, autophagy has dual roles in inflammation-driven pancreatic carcinogenesis. HMGB1, a DNA-binding protein and DAMP, promotes autophagy via multiple mechanisms.64Pancreatic Hmgb1 knockout (Pdx1-Cre;hmgb1^fl/fl) accelerates inflammatory damage, KRAS activation, and tumourigenesis more than Atg7 deletion.65Simultaneously, pancreatic cancer cells evade apoptosis the programmed cell death mechanism through the dysregulation of various pro- and anti-apoptotic signaling pathways. Despite accumulating genetic damage or being exposed to cytotoxic therapies, Because pro-apoptotic factors like Bax, p53, and caspases are downregulated or mutated, Even when anti-apoptotic proteins like Bcl-2 and Bcl-xL are overexpressed, these cells can still stop cell death.The simultaneous activation of autophagy and suppression of apoptosis thus creates a protective advantage for pancreatic cancer cells, fostering aggressive tumour behavior, resistance to therapy, and poor clinical outcomes. One possible tactic to improve therapy effectiveness and get past resistance in pancreatic cancer is to target these pathways with particular inhibitors or combination medicines.
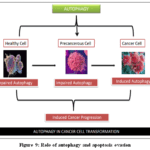 |
Figure 9: Role of autophagy and apoptosis evasion |
DNA damage repair pathways (ATM/ATR, BRCA)
The growth of tumours and resistance to treatment in pancreatic cancer are significantly influenced by the dysregulation of DNA damage repair (DDR) pathways, especially those involving the ATM/ATR and BRCA genes (Fig.10).The DNA damage response depends on the ataxia-telangiectasia mutant and ATM and Rad3-related kinases, which identify replication stress and DNA double-strand breaks, respectively, and trigger downstream effectors such CHK1 and CHK2 to stop the cell cycle and promote repair. Geographically, the incidence of PDAC varies significantly, with high-income countries having the highest frequency.66There are many known risk factors, both inherited and non-inherited, some of which could explain these differences, despite the fact that the causes of PDAC are intricate and multifaceted. Obesity, smoking, alcohol consumption, diabetes mellitus, chronic pancreatitis, and perhaps Helicobacter pylori infection are non-inherited risk factors.67Several genes are linked to increased PDAC susceptibility.68Germline DDR mutations (ATM, BRCA1, BRCA2, MLH1, MSH2, PALB2, PMS2, STK11) and traditional cancer risk genes (CDKN2A, TP53) are commonly inherited.69Some inherited cancer predisposition syndromes feature monoallelic, dominantly inherited mutations (e.g., BRCA1/2) that elevate PDAC and other cancer risks.70Mutations or functional deficiencies in ATM are observed in a subset of pancreatic ductal adenocarcinoma (PDAC) cases, leading to genomic instability and aggressive tumour phenotypes. Similarly, BRCA1 and BRCA2, key mediators of homologous recombination repair, are frequently mutated in hereditary and sporadic pancreatic cancers. Loss of BRCA function impairs high-fidelity DNA repair, making tumour cells more dependent on alternative, error-prone pathways, thereby promoting mutagenesis. While such deficiencies can enhance tumour aggressiveness, they also create therapeutic vulnerabilities particularly to platinum-based chemotherapy and PARP inhibitors through synthetic lethality. However, activation of compensatory DDR pathways, such as ATM/ATR signaling in BRCA-deficient cells, can confer resistance to these therapies, highlighting the complex interplay of DDR mechanisms in pancreatic cancer and the need for tailored combination strategies.
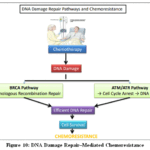 |
Figure 10: DNA Damage Repair–Mediated Chemoresistance |
miRNAs and non-coding RNAs influencing resistance
Through their effects on gene expression networks related to drug metabolism, apoptosis, epithelial–mesenchymal transition (EMT), and DNA repair, In pancreatic cancer, chemoresistance is regulated by microRNAs (miRNAs) and other non-coding RNAs (ncRNAs).Tumour suppressors or oncogenic miRNAs (oncomiRs) are two possible functions of dysregulated miRNAs.For instance, by targeting tumoursuppressor genes like PTEN and PDCD4, miR-21, which is commonly overexpressed in pancreatic ductal adenocarcinoma (PDAC), increases resistance to gemcitabine while improving cell survival and lowering apoptosis.Similarly, miR-155 and miR-221 have been linked to resistance by modulating pathways like NF-κB and PI3K/AKT. On the other hand, by promoting EMT and suppressing pro-apoptotic signaling, downregulation of tumour-suppressive miRNAs like miR-34a and miR-200 family lowers susceptibility to chemotherapeutics. A longer mRNA-like transcript is processed into the mature, active 19–24 nucleotide miRNA (Fig. 1). Most initial transcripts are transcribed by RNA polymerase II.71,72The pri-miRNA may contain pre-miRNA hairpins for multiple miRNAs, some clustered within related genes.73Transcription can be driven by a distal host gene promoter or a proximal miRNA promoter.71,72After nuclear cleavage by the DROSHA-containing microprocessor complex, pre-miRNA is exported to the cytoplasm via XPO5.72The final mature RNA molecule is put into the miRNA-induced silencing complex (miRISC), which contains ARGONAUTE, and becomes functionally active after being cleaved by another RNAse III DICER in the cytoplasm.Through its binding to partially complementary regions, usually on the 3′ UTR, miRNA guides miRISC to target mRNAs, reducing protein production via mRNA degradation, cleavage, or translational inhibition through interactions with mRNA-bound complexes like CCR4-NOT and eIF4A-G.71,72The seed region, which consists of the second to eighth nucleotides of the miRNA, is typically responsible for miRNA binding to the mRNA. This allows tens or even hundreds of mRNAs to interact with a single miRNA. The amount of protein synthesis reduction for each target gene depends on the number of binding sites, binding affinity, and relative abundance of miRNA to target mRNA.74By serving as molecular sponges for miRNAs or by directly interacting with signalling proteins, Treatment resistance in PDAC is also influenced by circular RNAs and long non-coding RNAs.For example, by activating the Wnt/β-catenin and AKT pathways, lncRNA HOTAIR and lncRNA PVT1 increase resistance to gemcitabine.Certain circRNAs, like circHIPK3, sequester tumour-suppressive miRNAs, indirectly activating pro-survival genes. Collectively, the dysregulation of these ncRNAs alters multiple signaling cascades, enabling pancreatic cancer cells to evade cytotoxicity. One promising therapeutic approach to combat chemoresistance in PDAC is to target certain miRNAs or lncRNA–miRNA interactions.
Comparative Analysis
Three commonly utilised human pancreatic cancer cell lines, each exhibiting distinct biological and molecular traits. PANC-1 and MiaPaCa-2, both harboring KRAS mutations, display mesenchymal-like phenotypes and higher drug resistance, with longer doubling times for PANC-1. In contrast, BxPC-3 is KRAS wild-type, more epithelial in nature, and generally more drug-sensitive. These differences make them valuable models for studying pancreatic cancer heterogeneity, drug responses, and specific pathways in tumour biology (Table 2).
Table 2: Comparative Characteristics of Commonly Used Pancreatic Cancer Cell Lines
| Cell Line | Origin | KRAS Status | Doubling Time (in vitro) | EMT / Phenotype | Drug Resistance Highlights |
| PANC-1 | Primary pancreatic ductal adenocarcinoma | KRAS-mutant (e.g., G12D) | ~52 h | Partial EMT; mesenchymal traits (↑vimentin,↓E-cadherin) | Intrinsic gemcitabine resistance (low hENT1, survival pathway activation); used to study EMT- and stemness-linked resistance.75 |
| MiaPaCa-2 | Pancreatic carcinoma patient derived (metastasis) | KRAS-mutant (e.g., G12C) | ~40 h | Mesenchymal-like; high plasticity, glycolytic | Resistant to gemcitabine; shows metabolism-, stemness-, and apoptosis-related resistance mechanisms.76 |
| BxPC-3 | Moderately differentiated pancreatic adenocarcinoma | KRAS wild-type | ~48–60 h | Epithelial phenotype (high E-cadherin, tight junctions) | Initially gemcitabine-sensitive; develops resistance via EMT, CXCR4/CXCR7 signaling, and anti-apoptotic pathways.77 |
Applications in Drug Screening
Cell lines serve as indispensable platforms for evaluating new therapeutic agents in pancreatic cancer research. Their unique genetic profiles such as variations in KRAS, TP53, SMAD4, and CDKN2A allow researchers to capture the heterogeneity of pancreatic tumours, making them ideal for high-throughput screening (HTS) platforms where thousands of compounds can be rapidly tested for cytotoxicity, apoptosis induction, and signaling pathway modulation.78Both natural products, such as plant-derived flavonoids and marine metabolites, and synthetic molecules, including kinase inhibitors and chemotherapeutic analogs, have been extensively screened using these models.79Combination therapy approaches, such as pairing standard agents like gemcitabine with novel targeted inhibitors, are frequently assessed in these cell lines to explore synergistic effects and overcome intrinsic or acquired drug resistance.80To better replicate the in vivo tumour microenvironment, these cell lines are increasingly cultured in 3D spheroid and organoid systems, They offer improved drug penetration profiles, nutrition gradients, and cell–matrix and cell–cell interactions, making them more accurate predictors of therapeutic outcomes.81Additionally, advanced molecular tools such as CRISPR/Cas9 genome editing and siRNA knockdown are employed to selectively silence or modify genes of interest. This facilitates the validation of novel drug targets, identification of resistance-conferring pathways, and elucidation of mechanisms underlying variable drug responses.82Collectively, these applications position cell lines as critical preclinical models for streamlining the drug discovery pipeline and translating laboratory findings into effective therapeutic strategies for pancreatic cancer.
Limitations And Future Perspectives
While cell lines have significantly advanced our understanding of pancreatic cancer biology and drug resistance mechanisms, they are not without limitations. A primary drawback lies in their reliance on conventional 2D monolayer culture systems, which fail to accurately mimic the complex architecture, extracellular matrix interactions, and heterogeneous tumour microenvironment of in vivo pancreatic tumours.83This oversimplification can lead to discrepancies in drug sensitivity and therapeutic response compared to clinical outcomes. To address these shortcomings, co-culture systemswhich integrate pancreatic cancer cells with stromal, immune, and endothelial components have emerged as powerful tools to recreate more physiologically relevant interactions. Similarly, organoid models derived from these lines or from patient samples can preserve tumour heterogeneity, genetic mutations, and spatial organization, providing a more accurate platform for translational research.84Beyond in vitro methods, researchers may now examine tumour growth, metastasis, and medication resistance under more clinically relevant circumstances thanks to patient-derived xenografts (PDXs) using these cells in immunodeficient mice. These models bridge the gap between cell culture and patient trials, offering valuable insights into tumour behavior in a living organism.85Looking ahead, the integration of these well-characterized pancreatic cancer cell lines into personalized therapy models is a promising avenue. By combining genomic profiling, drug response data, and advanced culture systems such as microfluidic chips and AI-driven analytics, researchers can tailor treatment strategies to individual patient profiles.86This multi-platform approach holds potential for improving drug discovery, refining therapeutic regimens, and ultimately enhancing clinical outcomes in pancreatic cancer management.
Conclusion
PANC-1, MiaPaCa-2, and BxPC-3 serve as cornerstone models in pancreatic cancer research, offering complementary insights into tumour heterogeneity, signaling pathways, and phenotypic variations. Their distinct molecular profiles and drug sensitivity patterns provide a robust foundation for dissecting the mechanisms underlying chemoresistance, particularly in the context of KRAS-driven and KRAS-independent pathways. However, given the complexity of pancreatic cancer biology, integrating these traditional cell line models with advanced platforms such as AI-driven predictive analytics, organ-on-chip systems, and patient-derived organoids will enhance translational relevance, accelerate drug discovery, and improve therapeutic precision.
Acknowledgement
The authors express their thanks to Saveetha College of Pharmacy – SIMATS for providing the necessary facilities to carry out this research work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval .
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials
Permission to reproduce material from other sources
Not applicable
Author Contributions
- Binoy Varghese Cheriyan: Visualization, Supervision, Project Admininstration.
- Kaniga Pandi: Writing – Original Draft.
- Kunal Rupesh Kumar Kataria: Funding Acquisition,
- Yokesh Shanmugam, Maha swethakannuchamy: Resources.
- Mahalakshmi sundarraj, Kaviyajothi Arumugasamy, Kaviyapriya Gopal Kuberaselvam: Data Collection.
- Praveen Kumar Ramesh, Bhuvaneshraj: Analysis.
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394-424. doi:10.3322/caac.21492
CrossRef - GBD 2017 Pancreatic Cancer Collaborators. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2019;4(12):934-947. doi:10.1016/S2468-1253(19)30347-4
CrossRef - Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an update. Dig Dis. 2010;28(4-5):645-656. doi:10.1159/000320068
CrossRef - Midha S, Chawla S, Garg PK. Modifiable and non-modifiable risk factors for pancreatic cancer: A review. Cancer Lett. 2016;381(1):269-277. doi:10.1016/j.canlet.2016.07.022.
CrossRef - Kaviyaprabha R, Miji TV, Sreelakshmi PS, Muthusami S, Arulselvan P, Bharathi M. Unveiling the Potential Role of Hesperetin and Emodin as a Combination Therapy to Inhibit the Pancreatic Cancer Progression against the C-Met Gene. Protein & Peptide Letters. 2025.
CrossRef - Sung H, Siegel RL, Rosenberg PS, Jemal A. Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. Lancet Public Health. 2019;4(3):e137-e147. doi:10.1016/S2468-2667(18)30267-6
CrossRef - Ntala C, Debernardi S, Feakins RM, Crnogorac-Jurcevic T. Demographic, clinical, and pathological features of early onset pancreatic cancer patients. BMC Gastroenterol. 2018;18(1):139. Published 2018 Sep 12. doi:10.1186/s12876-018-0866-z
CrossRef - Hashimoto D, Satoi S, Fujii T, et al. Is surgical resection justified for pancreatic ductal adenocarcinoma with distant abdominal organ metastasis? A position paper by experts in pancreatic surgery at the Joint Meeting of the International Association of Pancreatology (IAP) & the Japan Pancreas Society (JPS) 2022 in Kyoto. Pancreatology. 2023;23(6):682-688. doi:10.1016/j.pan.2023.07.005
CrossRef - Labori KJ, Katz MH, Tzeng CW, et al. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma – A population-based cohort study. Acta Oncol. 2016;55(3):265-277. doi:10.3109/0284186X.2015.1068445
CrossRef - Burris 3rd HA, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of Clinical Oncology. 2023;41(36):5482-92.
CrossRef - Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817-1825. doi:10.1056/NEJMoa1011923
CrossRef - Kang J, Hwang I, Yoo C, et al. Nab-paclitaxel plus gemcitabine versus FOLFIRINOX as the first-line chemotherapy for patients with metastatic pancreatic cancer: retrospective analysis. Invest New Drugs. 2018;36(4):732-741. doi:10.1007/s10637-018-0598-5
CrossRef - Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med. 2018;379(25):2395-2406. doi:10.1056/NEJMoa1809775
CrossRef - Strimpakos AS, Syrigos KN, Saif MW. Translational research in pancreatic cancer. Highlights from the “2011 ASCO Gastrointestinal Cancers Symposium”. San Francisco, CA, USA. January 20-22, 2011. JOP. 2011;12(2):120-122. Published 2011 Mar 9.
- Yoshida J, Ishikawa T, Endo Y, Matsumura S, Ohta T, Mizushima K, Hirai Y, Okayama T, Sakamoto N, Inoue K, Katada K. Metformin inhibits epithelial-mesenchymal transition in human pancreatic cancer cell lines. Cancer Research. 2019;79(13_Supplement):1038-.
CrossRef - Gradiz R, Silva HC, Carvalho L, Botelho MF, Mota-Pinto A. MIA PaCa-2 and PANC-1 – pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci Rep. 2016;6:21648. Published 2016 Feb 17. doi:10.1038/srep21648.
CrossRef - Shichi Y, Sasaki N, Michishita M, et al. Enhanced morphological and functional differences of pancreatic cancer with epithelial or mesenchymal characteristics in 3D culture. Sci Rep. 2019;9(1):10871. Published 2019 Jul 26. doi:10.1038/s41598-019-47416-w
CrossRef - Principe DR, Diaz AM, Torres C, et al. TGFβ engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene. 2017;36(30):4336-4348. doi:10.1038/onc.2016.500
CrossRef - Safa AR. Epithelial-mesenchymal transition: a hallmark in pancreatic cancer stem cell migration, metastasis formation, and drug resistance. J Cancer Metastasis Treat. 2020;6:36. doi:10.20517/2394-4722.2020.55
CrossRef - Kawase T, Yasui Y, Nishina S, et al. Fibroblast activation protein-α-expressing fibroblasts promote the progression of pancreatic ductal adenocarcinoma. BMC Gastroenterol. 2015;15:109. Published 2015 Sep 2. doi:10.1186/s12876-015-0340-0.
CrossRef - Su CC. Tanshinone IIA can inhibit MiaPaCa‑2 human pancreatic cancer cells by dual blockade of the Ras/Raf/MEK/ERK and PI3K/AKT/mTOR pathways. Oncol Rep. 2018;40(5):3102-3111. doi:10.3892/or.2018.6670.
CrossRef - Arora N, Liang H, Reed A, Chang JT, Cravatt BF, Zhou Y. Targeting lipid metabolism disrupts KRAS oncogenesis in pancreatic cancer. Cancer Research. 2024;84(6_Supplement):1655-.
CrossRef - Du Y, Zhang J, Tao Z, et al. Aloe emodin exerts potent anticancer effects in MIAPaCa-2 and PANC-1 human pancreatic adenocarcinoma cell lines through activation of both apoptotic and autophagic pathways, sub-G1 cell cycle arrest and disruption of mitochondrial membrane potential (ΛΨm). J BUON. 2019;24(2):746-753.
- Du J, Wang J, Tan G, et al. Aberrant elevated microRNA-146a in dendritic cells (DC) induced by human pancreatic cancer cell line BxPC-3-conditioned medium inhibits DC maturation and activation. Med Oncol. 2012;29(4):2814-2823. doi:10.1007/s12032-012-0175-2
CrossRef - Chen RY, Xu B, Chen SF, et al. Effect of oridonin-mediated hallmark changes on inflammatory pathways in human pancreatic cancer (BxPC-3) cells. World J Gastroenterol. 2014;20(40):14895-14903. doi:10.3748/wjg.v20.i40.14895
CrossRef - Zhu L, Qin H, Li PY, et al. Response gene to complement-32 enhances metastatic phenotype by mediating transforming growth factor beta-induced epithelial-mesenchymal transition in human pancreatic cancer cell line BxPC-3. J Exp Clin Cancer Res. 2012;31(1):29. Published 2012 Mar 29. doi:10.1186/1756-9966-31-29
CrossRef - Metildi CA, Kaushal S, Hoffman RM, Bouvet M. In vivo serial selection of human pancreatic cancer cells in orthotopic mouse models produces high metastatic variants irrespective of Kras status. J Surg Res. 2013;184(1):290-298. doi:10.1016/j.jss.2013.03.049
CrossRef - Oliverius M, Flasarova D, Mohelnikova-Duchonova B, et al. KRAS pathway expression changes in pancreatic cancer models by conventional and experimental taxanes. Mutagenesis. 2019;34(5-6):403-411. doi:10.1093/mutage/gez021
CrossRef - Henry KE, Dacek MM, Dilling TR, et al. A PET Imaging Strategy for Interrogating Target Engagement and Oncogene Status in Pancreatic Cancer. Clin Cancer Res. 2019;25(1):166-176. doi:10.1158/1078-0432.CCR-18-1485
CrossRef - Kim JB, Islam S, Kim YJ, et al. N-Cadherin extracellular repeat 4 mediates epithelial to mesenchymal transition and increased motility. J Cell Biol. 2000;151(6):1193-1206. doi:10.1083/jcb.151.6.1193
CrossRef - Karayiannakis AJ, Syrigos KN, Polychronidis A, Simopoulos C. Expression patterns of alpha-, beta- and gamma-catenin in pancreatic cancer: correlation with E-cadherin expression, pathological features and prognosis. Anticancer Res. 2001;21(6A):4127-4134.
- Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127(6 Pt 2):2021-2036. doi:10.1083/jcb.127.6.2021
CrossRef - El-Hariry I, Pignatelli M, Lemoine NR. FGF-1 and FGF-2 regulate the expression of E-cadherin and catenins in pancreatic adenocarcinoma. Int J Cancer. 2001;94(5):652-661. doi:10.1002/ijc.1515
CrossRef - Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148(4):779-790. doi:10.1083/jcb.148.4.779
CrossRef - Hirai I, Kimura W, Ozawa K, et al. Perineural invasion in pancreatic cancer. Pancreas. 2002;24(1):15-25. doi:10.1097/00006676-200201000-00003
CrossRef - Redies C, Engelhart K, Takeichi M. Differential expression of N- and R-cadherin in functional neuronal systems and other structures of the developing chicken brain. J Comp Neurol. 1993;333(3):398-416. doi:10.1002/cne.903330307
CrossRef - Spivakov M, Fisher AG. Epigenetic signatures of stem-cell identity. Nat Rev Genet. 2007;8(4):263-271. doi:10.1038/nrg2046
CrossRef - Alvarez CV, Garcia-Lavandeira M, Garcia-Rendueles ME, et al. Defining stem cell types: understanding the therapeutic potential of ESCs, ASCs, and iPS cells. J Mol Endocrinol. 2012;49(2):R89-R111. Published 2012 Aug 30. doi:10.1530/JME-12-0072
CrossRef - Mitsui K, Tokuzawa Y, Itoh H, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113(5):631-642. doi:10.1016/s0092-8674(03)00393-3
CrossRef - Tang F, Barbacioru C, Bao S, et al. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell. 2010;6(5):468-478. doi:10.1016/j.stem.2010.03.015
CrossRef - Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. cell. 2006 Aug 25;126(4):663-76.
CrossRef - Pera MF, Tam PP. Extrinsic regulation of pluripotent stem cells. Nature. 2010 Jun 10;465(7299):713-20.
CrossRef - Wen J, Park JY, Park KH, Chung HW, Bang S, Park SW, Song SY. Oct4 and Nanog expression is associated with early stages of pancreatic carcinogenesis. Pancreas. 2010 Jul 1;39(5):622-6.
CrossRef - Lu Y, Zhu H, Shan H, Lu J, Chang X, Li X, Lu J, Fan X, Zhu S, Wang Y, Guo Q. Knockdown of Oct4 and Nanog expression inhibits the stemness of pancreatic cancer cells. Cancer letters. 2013 Oct 28;340(1):113-23.
CrossRef - Quint K, Tonigold M, Di Fazio P, Montalbano R, Lingelbach S, Rueckert F, Alinger B, Ocker M, Neureiter D. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. International journal of oncology. 2012 Dec 1;41(6):2093-102.
CrossRef - Wang X, Liu Q, Hou B, Zhang W, Yan M, Jia H, Li H, Yan D, Zheng F, Ding W, Yi C. Concomitant targeting of multiple key transcription factors effectively disrupts cancer stem cells enriched in side population of human pancreatic cancer cells. PloS one. 2013 Sep 11;8(9):e73942.
CrossRef - Xu Q, Wang Z, Chen X, et al. Stromal-derived factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural invasion in pancreatic cancer. Oncotarget. 2015;6(7):4717-4732. doi:10.18632/oncotarget.3069
CrossRef
- Wang Z, Ma Q, Liu Q, et al. Blockade of SDF-1/CXCR4 signalling inhibits pancreatic cancer progression in vitro via inactivation of canonical Wnt pathway. Br J Cancer. 2008;99(10):1695-1703. doi:10.1038/sj.bjc.6604745
CrossRef - Qin-hong XU, Lian-kang SU, CHENG L, Can-can ZH, Wei-kun QI, YAN B, Qing-yong MA, WANG Z. Chemokine receptor CXCR7 assists CXCR4 in modulating the directional metastasis and EMT of pancreatic cancer. Xi’an jiao tong da xuexuebao. Yi xue ban. 2019(3):374.
- Garcea G, Doucas H, Steward WP, Dennison AR, Berry DP. Hypoxia and angiogenesis in pancreatic cancer. ANZ J Surg. 2006;76(9):830-842. doi:10.1111/j.1445-2197.2006.03872.x
CrossRef - Büchler P, Reber HA, Büchler M, et al. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas. 2003;26(1):56-64. doi:10.1097/00006676-200301000-00010
CrossRef - Bannoura SF, Uddin MH, Nagasaka M, et al. Targeting KRAS in pancreatic cancer: new drugs on the horizon. Cancer Metastasis Rev. 2021;40(3):819-835. doi:10.1007/s10555-021-09990-2
CrossRef - Bulle A, Liu P, Seehra K, et al. Combined KRAS-MAPK pathway inhibitors and HER2-directed drug conjugate is efficacious in pancreatic cancer. Nat Commun. 2024;15(1):2503. Published 2024 Mar 20. doi:10.1038/s41467-024-46811-w
CrossRef - Ireland L, Santos A, Ahmed MS, et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016;76(23):6851-6863. doi:10.1158/0008-5472.CAN-16-1201
CrossRef - Zeng S, Pöttler M, Lan B, Grützmann R, Pilarsky C, Yang H. Chemoresistance in Pancreatic Cancer. Int J Mol Sci. 2019;20(18):4504. Published 2019 Sep 11. doi:10.3390/ijms20184504
CrossRef - Zhao B, Qin C, Li Z, et al. Multidrug resistance genes screening of pancreatic ductal adenocarcinoma based on sensitivity profile to chemotherapeutic drugs. Cancer Cell Int. 2022;22(1):374. Published 2022 Dec 1. doi:10.1186/s12935-022-02785-7
CrossRef - Adamska A, Elaskalani O, Emmanouilidi A, et al. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. AdvBiolRegul. 2018;68:77-87. doi:10.1016/j.jbior.2017.11.007
CrossRef - O’Driscoll L, Walsh N, Larkin A, et al. MDR1/P-glycoprotein and MRP-1 drug efflux pumps in pancreatic carcinoma. Anticancer Res. 2007;27(4B):2115-2120.
- Li J, Chen X, Kang R, Zeh H, Klionsky DJ, Tang D. Regulation and function of autophagy in pancreatic cancer. Autophagy. 2021;17(11):3275-3296. doi:10.1080/15548627.2020.1847462
CrossRef - Antonucci L, Fagman JB, Kim JY, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl AcadSci U S A. 2015;112(45):E6166-E6174. doi:10.1073/pnas.1519384112
CrossRef - Zhou X, Xie L, Xia L, et al. RIP3 attenuates the pancreatic damage induced by deletion of ATG7. Cell Death Dis. 2017;8(7):e2918. Published 2017 Jul 13. doi:10.1038/cddis.2017.313
CrossRef - Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, White E. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell. 2014;55(6):916-930. doi:10.1016/j.molcel.2014.07.019
CrossRef - Yang S, Imamura Y, Jenkins RW, et al. Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation. Cancer Immunol Res. 2016;4(6):520-530. doi:10.1158/2326-6066.CIR-15-0235
CrossRef - Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881-892. doi:10.1083/jcb.200911078
CrossRef - Kang R, Xie Y, Zhang Q, et al. Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer. Cell Res. 2017;27(7):916-932. doi:10.1038/cr.2017.51
CrossRef - Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394-424. doi:10.3322/caac.21492
CrossRef - Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371(11):1039-1049. doi:10.1056/NEJMra1404198
CrossRef - Roberts NJ, Norris AL, Petersen GM, et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016;6(2):166-175. doi:10.1158/2159-8290.CD-15-0402
CrossRef - Takai E, Yachida S, Shimizu K, et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget. 2016;7(45):74227-74235. doi:10.18632/oncotarget.12490
CrossRef - Nicolosi P, Ledet E, Yang S, et al. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019;5(4):523-528. doi:10.1001/jamaoncol.2018.6760
CrossRef - Bartel DP. Metazoan MicroRNAs. Cell. 2018;173(1):20-51. doi:10.1016/j.cell.2018.03.006
CrossRef - Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20(1):21-37. doi:10.1038/s41580-018-0045-7
CrossRef - Fromm B, Billipp T, Peck LE, et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu Rev Genet. 2015;49:213-242. doi:10.1146/annurev-genet-120213-092023
CrossRef - Sempere LF, Azmi AS, Moore A. microRNA-based diagnostic and therapeutic applications in cancer medicine. Wiley Interdiscip Rev RNA. 2021;12(6):e1662. doi:10.1002/wrna.1662
CrossRef - Madden ME, Sarras MP Jr. Morphological and biochemical characterization of a human pancreatic ductal cell line (PANC-1). Pancreas. 1988;3(5):512-528. doi:10.1097/00006676-198810000-00003
CrossRef - Sipos B, Möser S, Kalthoff H, Török V, Löhr M, Klöppel G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: towards the establishment of an in vitro research platform. Virchows Arch. 2003;442(5):444-452. doi:10.1007/s00428-003-0784-4
CrossRef
- Lu YY, Jing DD, Xu M, Wu K, Wang XP. Anti-tumor activity of erlotinib in the BxPC-3 pancreatic cancer cell line. World J Gastroenterol. 2008;14(35):5403-5411. doi:10.3748/wjg.14.5403
CrossRef - Pan CH, Otsuka Y, Sridharan B, et al. An unbiased high-throughput drug screen reveals a potential therapeutic vulnerability in the most lethal molecular subtype of pancreatic cancer. Mol Oncol. 2020;14(8):1800-1816. doi:10.1002/1878-0261.12743
CrossRef - Agarwal T, Manivannan HP, Veeraraghavan VP, Sankaran K, Francis AP. Selective plant alkaloids as potential inhibitors of PARP in pancreatic cancer-An in silico study. Indian Journal of Biochemistry and Biophysics (IJBB). 2023;60(7):555-66.
- Manivannan G, Nimithap S, Rathika R, Kumar AG. Intersection of Precision Medicine and Cancer Therapy.
- Frappart PO, Walter K, Gout J, et al. Pancreatic cancer-derived organoids – a disease modeling tool to predict drug response. United European Gastroenterol J. 2020;8(5):594-606. doi:10.1177/2050640620905183
CrossRef - Yasunaga K, Ito T, Miki M, et al. Using CRISPR/Cas9 to Knock out Amylase in Acinar Cells Decreases Pancreatitis-Induced Autophagy. Biomed Res Int. 2018;2018:8719397. Published 2018 May 17. doi:10.1155/2018/8719397
CrossRef - Suri R, Zimmerman JW, Burkhart RA. Modeling human pancreatic ductal adenocarcinoma for translational research: current options, challenges, and prospective directions. Ann Pancreat Cancer. 2020;3:17. doi:10.21037/apc-20-29
CrossRef - Schuth S, Le Blanc S, Krieger TG, et al. Patient-specific modeling of stroma-mediated chemoresistance of pancreatic cancer using a three-dimensional organoid-fibroblast co-culture system. J Exp Clin Cancer Res. 2022;41(1):312. Published 2022 Oct 22. doi:10.1186/s13046-022-02519-7
CrossRef
- Garcia PL, Miller AL, Yoon KJ. Patient-Derived Xenograft Models of Pancreatic Cancer: Overview and Comparison with Other Types of Models. Cancers (Basel). 2020;12(5):1327. Published 2020 May 22. doi:10.3390/cancers12051327.
CrossRef - Leroux C, Konstantinidou G. Targeted Therapies for Pancreatic Cancer: Overview of Current Treatments and New Opportunities for Personalized Oncology. Cancers (Basel). 2021;13(4):799. Published 2021 Feb 14. doi:10.3390/cancers13040799.
CrossRef
Abbreviations
PDAC – Pancreatic Ductal AdenoCarcinoma
5-FU – 5-fluorouracil
DFS – Disease-Free Survival
OS – Overall Survival
CRISPR – Clustered Regularly Interspaced Short Palindromic Repeats.
PANC-1 – Pancreatic carcinoma-1
MiaPaCa-2 – Miami Pancreatic Carcinoma-2
BxPC-3 – Boxer Pancreatic Carcinoma-3
KRAS –Kirsten Rat Sarcoma Viral Oncogene Homolog
MAPK –Mitogen-Activated Protein Kinase
PI3K – Phosphatidylinositol-3-Kinase
AKT- Protein Kinase B
mTOR- Mammalian Target of Rapamycin
HIF-1α – Hypoxia-Inducible Factor-1 alpha
VEGF – Vascular Endothelial Growth Factor
ATM –Ataxia Telangiectasia Mutated
ATR- ATM and Rad3-Related
BRCA- Breast Cancer Susceptibility Gene




