Structure-Based in Silico Insights of Sitagliptin Analogs Against Dipeptidyl Peptidase-4

Department of Pharmaceutical Chemistry, College of Pharmacy, University of Baghdad, Baghdad, Iraq
Corresponding Author E-mail: hala.abd@copharm.uobaghdad.edu.iq
DOI : http://dx.doi.org/10.13005/bpj/3450
Download this article as:
![]()
Dipeptidyl peptidase-4 DPP-4 inhibition has remained one of the most established ways in treating type 2 diabetes mellitus. Therefore, the development of improved DPP-4 inhibiters is consideredan essential strategy in the design of antidiabetic drugs. In the present study, a structure-based virtual approach was considered to design and evaluate new sitagliptin analogues with improved binding stability and pharmacokinetic properties. A virtual library consisting of 15,034 different compounds was created and then screened using a hierarchical Glide docking protocol (HTVS, SP, and XP). Using integrated computational analyses, the top-ranked compounds were further assessed. Based on ADMET predictions, MM-GBSA binding free energy calculations, the results revealed that out of all the screened compounds, only 28 candidates had better docking scores than sitagliptin and compounds 14 and 22 were the most promising screened analogues. Compared to other tested analogies, compound 14 showed stable binding profile,low MM-GBSA binding free energy,stable ionic interactions between the catalytic residues Glu205 and Glu206, stable hydrophobic interactions within the S1 pocket. In addition, based on ADME simulation, compound 14 presented acceptable drug-like properties which indicates good oral bioavailability and lower predicted CNS penetration compared to sitagliptin. Overall, further experimental validation and development is required to evaluate compound 14 which exhibitsan increased electrostatic complementarity, dynamically stable properties, and acceptable pharmacokinetic properties making it a promising candidate as a DPP-4 inhibitor.
KEYWORDS:ADME; DPP-4 inhibitors; In silico design; MM-GBSA; Sitagliptin analogues
Introduction
Insulin resistance, combined with the gradual failure of β-cells, defines type2 diabetes mellitus (T2DM), a long-term metabolic disorder that disrupts glucose balance.The increasing prevalence of age-related metabolic disorders gives rise to an urgent need for targeted therapies to promote healthy aging among older adults. SinceT2DM comprises one of the most prevalent age-associated metabolic disorders worldwide, further comprehension of its molecular mechanisms and therapeutic targets is necessary to develop safer and more effective interventions. Glucagon-like peptide-1 GLP-1 and Glucose-dependent Insulinotropic Peptide GIP are two examples of the incretin hormones that are crucial for controlling blood sugar levels after meals. They help increase insulin release and decrease glucagon secretion in response to glucose.1,2
The serine protease dipeptidyl peptidase-4 (DPP-4) is primarily present in immune and endothelial cells. Its primary function is that it quickly breaks down these incretins.3As a result, the long-term therapeutic effectiveness of GLP-1 and GIP is significantly limited due to their short lifetime.Inhibiting DPP-4 extends the action of the incretins, which in turn supports insulin release and improves blood sugar control.4 This principle provides a basis for using “DPP-4 inhibitors gliptins”, a well-known class of oral diabetes drugs.5
The first DPP-4 inhibitor to be clinically effective was sitagliptin, which was approved in 2006 and has been a crucial model for the development of more recent medications.6The triazolopiperazine structure shows a strong attraction to the DPP-4 active site, interacting with the S1 and S2 binding pockets. Later generations of DPP-4 inhibitors can produce a variety of analogues because of this interaction. 7,8The understanding of this binding architecture has facilitated the investigation of modified chemicals necessary for better potency and selectivity. Over the past ten years, computer-based methods have become crucial tools for developing and improving DPP-4 inhibitors. Researchers can accurately predict binding affinity and assess large collections of chemicals using methods like molecular docking, structure-based virtual screening, and molecular dynamics simulations.9These methods have uncovered new structures and derivatives from natural sources that show strong inhibition against DPP-4.10,11
Quantitative Structure-Activity Relationship QSAR and Structure-Activity Relationship SARmodeling assist medicinal chemists in identifyingsignificant molecular features that immensely influence both effectiveness and selectivity.12Recently, to enhance the prediction performance,researchers have investigated the integration of machine learning ML and artificial intelligence AIwith conventional docking techniques. Such integration assistsin reducing time and experimental cost by allowing automated hit prioritization and data- driven analogue generation.Neuro-symbolic and deep learning approaches have shown high accuracy in predicting active DPP-4 inhibitors, thus lowering the experimental workload.13,12
The successful implementation of computer-based simulations,commonly known as in silico techniques, has demonstrated that it can play a pivotal role in all drug discovery stages from hit finding to SAR optimization. Researchers can systematically and rapidly develop compounds such as sitagliptin.15,16As a result, these techniques provide a fundamental strategy necessary for designing sitagliptin-derived scaffolds with enhanced pharmacodynamic and pharmacokinetic properties.However, despite the beneficial therapeutical effect of thefirst-generation of gliptinssuch as sitagliptin, significant limitations still exist.Post-marketing and clinical data have reported several rare but dangerous side effects, such as severe joint pain, acute pancreatitis, and bullous pemphigoid related to immunological mechanisms.17,18,19Sitagliptin has a limited metabolism and is primarily eliminated unaltered through urine (roughly 79–87% of an oral dose). This fact restricts its utilization forpatients with severe renal impairment and thereby dose adjustment is required for those who have kidney functioning problems.20Furthermore, its broad distributionmay reduce tissue selectivity and lead to negative effects on theoff-target regions such asthe skin and musculoskeletal system.21
In recent years, there has been significant progress in the structural optimization of DPP-4 inhibitors through computational chemistry techniques. Various studies have shown that the application of docking and molecular dynamics methods together enhances the accuracy of DPP-4 inhibitory potency and selectivity.22,23In addition, artificial intelligence-assisted screening platforms have accelerated hit identification while reducing synthetic burden.These technological advancements facilitate the transition from empiricaldrug development towardmolecularlyderived design techniques.24,25
Clinical safety studies and pharmacovigilance approaches have also shown that there is a need to optimize pharmacokinetics and immunology while developing second-generation gliptins.26,27All these studies together emphasize the need to design rationally active sitagliptin analogs with high binding efficiency and better peripheral selectivity. is highlighted by these safety and pharmacokinetic concerns.
The aim of the current investigation is to design and simulate new sitagliptin-based analogues using SAR-guided in silico techniques, particularlymultistep docking, ADMET predictions, and MM-GBSA free energy calculations.The purpose of the designed compounds is to increase tissue selectivity and minimize off-target actions. The chemical structure of the suggested compounds is illustrated in Figure 1.
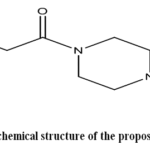 |
Figure 1: The chemical structure of the proposed compounds. |
Materials and Methods
Computational Method
In this section, the computational methods that were used to design and simulate new sitagliptin-based analogues will be presented.Molecular docking studies were conducted using Schrodinger suite tools on Maestro (version 14.5.160, 2025-3; Schrodinger LLC, New York, NY, 2025), including Glide, Desmond, and Qikprop. The computing facility was provided by the University of Baghdad’s College of Pharmacy’s Departmentof Pharmaceutical Chemistry.
Molecular Docking
Ligand structures were prepared using the LigPrep module. The Protein Preparation Wizard was employed to process the crystal structures for DPP-4/CD26 (PDB ID: 2P8S), which was obtained from the Protein Data Bank.28
ADME Study
To predict the drug likeness characteristics of the proposed molecules, the ADMET properties including absorption, distribution, metabolism, and excretionof the proposed ligands were evaluated using the QikProp program. Molecular weight (MW), number of hydrogen-bond donors (donaorHB), number of hydrogen-bond acceptors (accprHP), predicted apparent Caco-2 cell permeability (QPPCaco), and predicted human oral absorption (oralAbs) as suggested in.29
MM/GBSAStudy
MM-GBSA study was performed on the complexes ofthe protein–ligand that were produced by molecular docking against DPP-4/CD26 (PDB 2P8S). In the investigations, the total binding free energy (ΔG_bind, kcal mol⁻¹) of each ligand and its decomposed energy componentsvan der Waals (vdW), Coulombic (electrostatic), and generalized-Born solvation (GB)were computed. The calculated ΔG_bind provides a trustworthy internal ranking based on the balance of hydrophobic packing, electrostatics, and desolvation penalties due to the identical utilization of the receptor and grid for all ligands.30
Molecular Dynamics Simulation
To assess the dynamic stability of the complexes, the top-ranked molecule from the docking studies wasfurther investigated using the Desmond module.31,32BothRoot Mean Square Fluctuation (RMSF) and Root Mean Square Deviation (RMSD) values were computed of the backbone atoms in the receptor protein and the protein-ligand complex.33
Results
Molecular Docking analysis
In the present investigation, a library comped of 15034 compounds wasfirstly screened against DPP-4 (CD26) enzyme using high throughput virtual screening (HTVS) molecular docking. After that, these compounds were initially re-docked with Standard Precision (SP) followed by ExtraPrecision (XP) to optimize screening accuracy and obtain more reliablepredictions. The workflow that was followed in the molecular modeling study is illustrated in Figure 2. Because of their low docking energiescompared to the sitagliptin, 16 compounds obtained from XP screening were chosen for further analysis, as demonstrated in Table 1.
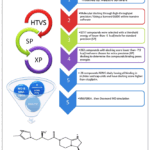 |
Figure 2: The steps for molecular modeling studies. |
 |
Table 1: Docking scores of the proposedcompounds on the protein (2P8S) |
ADME Study
Because of the crucial role of Pharmacokinetic in predicting absorption, distribution, and metabolism in drug design, the therapeutic effectiveness of the proposed compounds was investigated. The Pharmacokinetic properties of the top-ranking molecules were predicted using the QikProp module that is available in Schrödinger Suite. Thesimulation data are summarized in Table 2.
The results showed that the molecular weights of all designed analogues varied between 290 and 315 Da, whichare far below the Lipinski’s Rule of Five threshold. All the compounds showed good human oral absorption and acceptable HBD and HBA counts. This indicates that the proposed compounds support Rules of Three and Five.In addition, the results showed thatmost of the molecules had low-medium to high QPPCaco. This indicates good intestinal permeability and supports the potential for oral delivery. Furthermore, most compounds were expected to be non-CNS active (CNS from −2 to 0), indicating low potential for central side effects.
Among analyzed derivatives, compound 14 exhibited the most equally balanced pharmacokinetic profile, with QPPCaco of 23.66 nm/s, no rule violation and with CNS = −2 moderate permeability, good oral absorption and peripheral selectivity. Similarly, compound 22 also presented a favorable absorption (QPPCaco = 35.65 nm/s) and it was in accordance with drug-likeness rules; with slight difference in its neutral CNS score (CNS = 0). Although, compound 6 showed a desirable profile (QPPCaco = 26.41 nm/s) and complete rule compliance in the analysis, it slightly provides more predicted metabolic routes,which might affect stability as well. On the other hand, the results showed that compound 7 showed comparable permeability (28.65 nm/s) but weaker binding kinetics, making it a less potent albeit pharmacokinetically acceptable lead. In contrast, compound 16 exhibited a higher permeability (QPPCaco = 84.81 nm/s) yet positive CNS activity which could be used as an indicator of being a potentially CNS-penetrant molecule with off-target effects. The reference drug sitagliptin (QPPCaco of 60.05 nm/s and CNS = +1) validated the high permeability of the molecule with a potential to penetrate blood–brain barrier as an observation of its systemiceffects were reported.From the ADME results one can notice that of these analogs,compounds 14 and 22 showed improved PK and safety profiles as compared to sitagliptin, which confirm their potential as better DPP-4 inhibitors. Therefore, these two compounds were chosen for further assessmentusingin silico and in vitro studies.
Table 2: Pharmacokinetic properties for the top-ranking compounds.
| Comp. ID | mol MW | CNS | donorHB | accptHB | HumanOral
Absorption |
QPPCaco | #metab | Rule of Three |
| 1 | 307.35 | 1 | 2.000 | 7.700 | 2 | 7.475 | 8 | 2 |
| 2 | 307.35 | -2 | 4.000 | 8.200 | 1 | 2.650 | 10 | 2 |
| 3 | 307.35 | -2 | 4.000 | 8.200 | 1 | 3.594 | 10 | 2 |
| 4 | 310.37 | -1 | 3.000 | 7.450 | 2 | 40.950 | 8 | 1 |
| 5 | 305.33 | -2 | 3.000 | 8.950 | 2 | 19.141 | 9 | 2 |
| 6 | 307.35 | -2 | 2.000 | 8.700 | 2 | 26.406 | 6 | 0 |
| 7 | 295.29 | -2 | 2.000 | 9.700 | 2 | 28.648 | 5 | 0 |
| 8 | 307.35 | 1 | 2.000 | 7.700 | 2 | 10.227 | 8 | 2 |
| 9 | 307.35 | 1 | 2.000 | 7.700 | 2 | 10.335 | 8 | 2 |
| 10 | 308.33 | -2 | 3.000 | 8.200 | 2 | 24.862 | 7 | 1 |
| 11 | 293.32 | -1 | 3.000 | 10.200 | 2 | 5.958 | 6 | 1 |
| 12 | 306.36 | -2 | 5.000 | 7.700 | 1 | 2.149 | 9 | 2 |
| 13 | 308.33 | -2 | 2.000 | 9.450 | 2 | 18.316 | 7 | 2 |
| 14 | 306.36 | -2 | 2.000 | 8.700 | 2 | 23.661 | 6 | 0 |
| 15 | 306.367 | -1 | 5.000 | 7.700 | 2 | 7.596 | 9 | 2 |
| 16 | 312.361 | 1 | 2.000 | 6.700 | 2 | 84.811 | 7 | 1 |
| 17 | 307.352 | 1 | 2.000 | 7.700 | 2 | 13.421 | 8 | 2 |
| 18 | 307.352 | 0 | 2.000 | 8.200 | 2 | 50.171 | 7 | 1 |
| 19 | 290.324 | -2 | 2.000 | 9.200 | 2 | 31.096 | 8 | 1 |
| 20 | 307.352 | 0 | 2.000 | 6.700 | 2 | 40.494 | 7 | 1 |
| 21 | 292.337 | 1 | 2.000 | 7.200 | 2 | 90.126 | 7 | 1 |
| 22 | 306.367 | 0 | 2.000 | 8.200 | 2 | 35.655 | 6 | 0 |
| 23 | 312.755 | 1 | 2.000 | 7.200 | 2 | 90.551 | 7 | 1 |
| 24 | 307.352 | -1 | 4.000 | 8.200 | 1 | 4.633 | 10 | 2 |
| 25 | 312.361 | 1 | 2.000 | 6.700 | 2 | 87.113 | 7 | 1 |
| 26 | 306.367 | -2 | 5.000 | 7.700 | 1 | 2.592 | 9 | 2 |
| 27 | 307.352 | 1 | 2.000 | 7.700 | 2 | 12.422 | 8 | 2 |
| 28 | 308.336 | 0 | 2.000 | 8.900 | 2 | 44.944 | 8 | 1 |
| Sitagliptin | 407.318 | 1 | 1.000 | 5.000 | 3 | 60.049 | 5 | 0 |
MM/GBSA Study
The MM-GBSA (Molecular Mechanics–Generalized Born Surface Area) was used to post-process the results of docking and to estimate the binding free energies for selected compounds with promising activities, i.e. Compounds14,22, 6, 16,7, and reference sitagliptin Binding free energy and its decomposition of the calculated parameters are listed in Table 3, which consists of the total binding free energy (ΔG_bind) and decomposed energetic terms (i.e. van der Waals, Coulombic, lipophilic intermolecular contacts and solvent contribution).
The analysis revealed that all the designed compounds showed lower binding free energies than the effective compound sitagliptin (−50.69 kcal mol⁻¹), which confirmed stronger predicted affinities toward the DPP-4 active site. Of these tested compounds, compounds 14 and 22with total binding free energy (ΔG = −76.48 kcal mol⁻¹) and (ΔG =−76.46 kcal mol⁻¹), respectively exhibited the most stable binding characterized by optimal van der Waals and electrostatic contributions while being penalized with less desolation penalty. This interaction was also observed for compounds 6 and 16 but with larger solvation resistance, and relatively weakly in the case of compound 7. It can be said that compound 14 is the energetically most favored ligand based on MM-GBSA calculations followed closely bycompound 22 and the commercial compound sitagliptin in terms of predicted binding affinity and ligand efficiency.
Table 3: MM/GBSA Related Parameters forsitagliptin, compounds 14, 22, 6,16, and 7
| Compound | ΔG_bind (kcal/mol) | vdW | Coulomb | Lipo. intermolecular contacts | SolvGB | Ligand Efficiency |
| 14 | −76.48 | −36.27 | −66.16 | −16.70 | +42.76 | −3.48 |
| 22 | −76.46 | −19.29 | −93.56 | −14.14 | +46.23 | −3.47 |
| 6 | −68.39 | −28.05 | −101.87 | −15.72 | +75.76 | −3.10 |
| 16 | −67.20 | −28.08 | −82.14 | −15.85 | +69.80 | −3.09 |
| 7 | −53.95 | −25.61 | −91.10 | −12.80 | +71.99 | −2.57 |
| Sitagliptin | −50.69 | −33.83 | −56.07 | −19.38 | +62.11 | −1.81 |
ΔG_bind: Binding Free Energy; vdW: van der Waals Energy; Coulomb: Electrostatic (Coulombic) Energy; SolvGB: Generalized-Born Solvation Energy; pKᵢ (from ΔG): Negative Log of the Inhibition Constant.
Molecular Dynamic Simulation Study
Molecular dynamics (MD) simulations were used to assess dynamic aspects of the ligand-target interactions. Both Sitagliptin and the most active ligand (Compound 14) were considered for MD simulations to investigate the time dependent variation in their binding affinities towards receptor. These simulations explore how the ligands bind with critical protein residues in the binding pocket and so influence ligand occupancy and biological activity.
Protein-ligand RMSD value (PL-RMSD)
In this section, the protein ligand RMSD values are investigated, as can be seen in Figure 3. The complexes of Sitagliptin and compound 14 achieved equilibration over 100 ns, as evidenced by the plateau values in their RMSD plots and sustained fluctuations at 1–3 Å (structural stabilization), which indicatesa well-packed globular protein. Blue and red colors stand for receptor and ligand respectively. Sitagliptin protein RMSD saturated at 2.0 Å and ligand RMSD at 1.0 Å, indicating tight and stable binding, as illustrated Figure3. On the other hand,compound 14 presented a similar profile with the protein RMSD 2.2 Å and ligand RMSD of 1.3 Å, suggesting a slight increased mobility while keeping bound tight. One can also notice that in both tested cases, ligand RMSD was less than protein RMSD. Therefore, no liganddrifted from the pocket occurred and both complexes reached stable equilibrium. However, Sitagliptin showed slightly overall rigidity compared with compound 14.
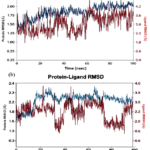 |
Figure 3: The RMSD values for (a)compound 14and (b) sitagliptin. |
Ligand RMSF (L-RMSF)
The ligand-RMSF gives detailed information about fluctuation level of the atom of ligand. This can be seen in the upper panel as it has a 2-dimensional structural illustration. The examination of L-RMSF further contributes to a clear elucidation in which ligand fragments engage in the interaction with protein and provide entropic contributions upon binding. In the present studies, a ligand predominantly showed L-RMSF values below 1.5 Å for DPP-4/CD26, suggesting solid stability inside binding sites.
In the result of the current L-RMSF analysis showed that in both ligands, the atomic displacements were low, which indicatesa stable binding in the active site. In addition, sitagliptin presented an average RMSF 0.5 Å, indicating subtle atomic motion particularly in the protonated amine and rigid bicyclic platform, and slight movement (< 1 Å) of the trifluorophenyl ring as well as side chains as shown in Figure 4. From Figure 4, compound 14 has a slightly higher RMSF (0.7 Å), which is an indication of moderately more flexible behavior. This can be attributed to the availability of multiple charged centers and rotatable bondsin compound 14. Both ligands were quite stable in the binding site, with Sitagliptin being better anchored and having lower internal reorganization than CPD-14.
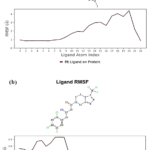 |
Figure 4: The MS simulation L-RMSF value for (a)Compound 14 and (b) Sitagliptin. |
Protein-RMSF (P-RMSF)
The Protein- RMSF(P-RMSF) provided results from the molecular dynamics simulation in a quantitative manner. It describes how much more rigid or flexible specific regions of the protein (residues) are relative to other parts over time. Well-defined regions such as alpha helixes and beta strands in general are less mobile than unstructured ones like loops.Both Sitagliptin and CPD-14 complexes presented very similar RMSF profiles, as illustrated in Figure 5. From Figure 5, it can be noticed that compound 14 has an average residue fluctuation of ca ~1.0 Å, which is an indication of the general stability of the protein. In addition, Low RMSF values within the α-helices and β-strands reflect relatively rigid secondary structures with higher peaks (> 1.5 Å) mainly observed at the N-terminus, C-terminus, and surface loops. Moreover, the active-site residues contacting each ligand and were consistently rigid for both complexes, indicating no disruption due to binding. Thenonhomogeneous behavioris consistent with the local flexibility ofcompound 14 bindingsite. In general, it was found that both ligands were equally well bounded to the DPP-4 protein with similar stabilized DPP-4 dynamic profiles during the simulation.
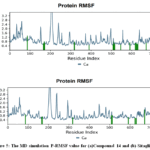 |
Figure 5: The MD simulation P-RMSF value for (a)Compound 14 and (b) Sitagliptin. |
Protein-Ligand Contacts
Both Sitagliptin and compound 14 share two primary interactions to the DPP-4 active site ionic anchoring via their primary amine and aromatic pocket stabilization.Primary Amine (Ionic) Interactions:The NH3+ group of each ligand makes strong salt bridges and hydrogen bonds with Glu205 and Glu206, the catalytic pocket’s critical acidic residues. UnlikeSitagliptin, which consistently maintains these contacts, compound 14 has a stronger, higher occupancy interaction with the ionic partner, presumably due to its higher positive charge (+3) and potential for making multivalent salt bridges. This stronger ionic binding contributes to a higher proportion of simulation frames in which the compound 14 is anchored to, thus it increases the overall electrostatic stability afforded by the pocket.
Aromatic Ring Interactions: the aromatic ring of both ligands is in the active site pocket, interacting with Phe357 and Tyr662 through π–π interaction and hydrophobic contacts. The trifluoro phenyl ring of sitagliptin is capped by strong hydrophobic packing, whereas the compound 14 heteroaromatic ring forms a similarly stable interaction which could be reinforced by π–cation interactions with Tyr662, as presented inFigure 6.In conclusion, both ligands have a stable binding, but compound 14 has a tighter ionic network with Glu205/206 an ionic contacts. However, Sitagliptin shows equal hydrophobic interaction to slightly favorable aromatic ring interaction stabilization.
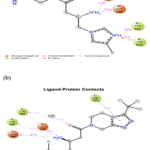 |
Figure 6: The interaction probability of (a) Compound 14 and (b) Sitagliptin. |
Discussion
A comprehensive structure-based computational analysis has led to identification of several proposed improved analogs of sitagliptin that will interact at the active site of DPP4. The data obtained via docking prior to any energetics/dynamics analyses is indicative of potential improved binding for several of the analogs; however, further energetic and dynamic analyses were required to identify if improvements in binding were a representative of true binding stability.
For compounds 14 and 22, binding scores consistently exceeded binding of sitagliptin through several tiers of analysis. In addition, with respect to improved scores associated with docking, MM-GBSA and free binding energies were also significantly lower for these compounds when compared to sitagliptin. Additionally, the differences in free binding energies (ΔG bind) were primarily attributable to greater electrostatic contributions as well as improved van der Waals interactions between the compounds and the catalytic pocket of DPP4, thereby providing for superior complementarity within the catalytic pocket of DPP4.
The primary mechanistic result from this study was that compound 14 formed a stronger ionic binding to Glu205 and Glu206 than did sitagliptin. Both compounds form stable salt bridges with these residues. However, in contrast to sitagliptin, compound 14 exhibited increased ionic persistence through molecular dynamics simulation, supporting improved charge distribution and enhancing stability of the active region. The increased occupancy of these ionic interactions likely facilitated the greatly improved free energy profile associated with these compounds.
The role of an aromatic chemical system in hydrophobic stabilization of the S1 pocket has been shown to be significant. For example, the 4,6-di(3,5-dimethyl-phenyl)-pyrimidin-2-amine series (compound 14) was found to provide stable, consistent π–π (aromatic) and hydrophobic interactions with Phe357 and Tyr662 that were comparable to or greater than those found with the dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin. This stability was attributed to the combination of stable electrostatic anchoring and stable hydrophobic packing of compound 14.
The pharmacokinetic predictions indicate that compound 14 has the optimal balance of permeability versus peripheral selectivity relative to sitagliptin. The negative central nervous system (CNS) score of compound 14 indicates less penetration of the blood brain barrier relative to sitagliptin,and therefore implies fewer central nervous system adverse effects. In contrast, compound 16, though predicted to be highly permeable, had a predicted CNS activity that may limit its safety profile.
In contrast to optimizing compounds through structural similarity-based methodology, our design approach has been to improve dynamic stability and minimize desolvation penalties. The reduction of the solvation energy penalty associated with compound 14 confirms greater relative binding efficiency.
Overall, the results from docking studies, MM-GBSA calculations, ADMET predictions and molecular dynamics modeling provide overlapping evidence that supports enhanced electrostatic complementarity, sustained interaction stability and improved predicted pharmacokinetic selectivity of compound 14 relative to sitagliptin.
Conclusion
The aim of the current investigation is to design new sitagliptin-based analogues using in silico simulation techniques. These simulations included multistep docking, ADMET predictions, MM-GBSA free energy calculationsand 100 ns MD simulations. A library of 15034 compounds was initially screened against DPP-4 (CD26) enzyme.The results revealed thatamong the screened sitagliptin analogs, compound 14 displayed the best overall profile. It showed the most favorable free binding energy and consistently formed ionic interactions with Glu205 and Glu206. In addition, there were stable hydrophobic contacts within the S1 pocket and reasonable predicted pharmacokinetics with reduced CNS penetration. On the other hand, Compound 22 showed promising characteristics as well. However, it exhibited lower energetic stability compared to Compound 14. Therefore, compound 14 can be considered as a lead compound for further investigation in vitro and in vivo experiments to prove its potency, selectivity and safety be carried out for their biological activity.
Acknowledgement
The author thanks the University of Baghdad’s College of Pharmacy for their assistance in providing the software needed for this analysis.
Funding Sources
For this article’s research, writing, and/or publication, the author did not receive any funding.
Conflicts of Interest
There are no conflicts of interest for the author.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to Reproduce Material from other Sources
Not Applicable
Author Contributions
The sole author was responsible for the conceptualization, methodology, data collection, analysis, writing, and final approval of the manuscript.
References
- Srivastava S, Haneef M, Saxena VL, Khan M, Khan S. Structure and ligand-based in silico studies towards the natural inhibitors against receptor recognition spike protein of SARS-CoV-2. Open Bioinform J. 2024;17:e18750362284177.
CrossRef - Deacon CF. Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2020;16(11):642-653.
CrossRef - Rizzo M, Nauck MA, Mantzoros CS. Incretin-based therapies in 2021—current status and future perspectives. Metabolism. 2021;122:154843.
CrossRef - Shao DW, Zhao LJ, Sun JF. Synthesis and clinical application of representative small-molecule DPP-4 inhibitors. Eur J Med Chem. 2024;272:116464.
CrossRef - Bayanati M, Mahboubi Rabbani MI, Kabiri SS, et al. DPP-4 inhibitors: a systematic review of structure–activity relationship studies. Iran J Pharm Res. 2024;23(1):e151581.
CrossRef - Saini K, Sharma S, Khan Y. DPP-4 inhibitors for treating T2DM—hype or hope? Front Mol Biosci. 2023;10:1130625.
CrossRef - Dutta R, Khatun S, Ghosh P, et al. Exploring DPP-4: implications in diseases and insights for inhibitor design. Bioorg Chem. 2025;165:108947.
CrossRef - Mobeen B, Shah M, Rehman HM, et al. Discovery of a selective and nanomolar inhibitor of DPP-4 more potent than sitagliptin. Eur J Med Chem. 2024;279:116834.
CrossRef - Zare F, Ataollahi E, Mardaneh P, et al. Virtual screening and molecular dynamics reveal potent and selective DPP-4 inhibitors. Sci Rep. 2024;14:7749.
CrossRef - Carpio LE, Olivares M, Ortega-Vallbona R, et al. DPPPRED-IV: an ensembled QSAR web server for DPP-4 inhibitors. Int J Mol Sci. 2025;26(12):5579.
CrossRef - Bustamam A, Hamzah H, Husna NA, et al. Artificial-intelligence paradigm for ligand-based virtual screening on type 2 diabetes drug discovery. J Big Data. 2021;8:74.
CrossRef - Hossain D, Saghapour E, Chen JY. NeSyDPP-4: discovering DPP-4 inhibitors with a neuro-symbolic AI approach. Front Bioinform. 2025;5:1603133.
CrossRef - Huang J, Jia Y, Sun S, Meng L. Adverse-event profiles of DPP-4 inhibitors: FDA pharmacovigilance data mining. BMC PharmacolToxicol. 2020;21(1):68.
CrossRef - Lee H, Chung HJ, Pawar A, et al. Risk of bullous pemphigoid with initiation of DPP-4 inhibitors. JAMA Dermatol. 2020;156(10):1107-1114.
CrossRef - Chanprapaph K, Pratumchart N, Limtong P, et al. DPP-4 inhibitor-related bullous pemphigoid in diabetic patients. J Dermatol. 2021;48(4):486-496.
CrossRef - Daza-Arnedo R, Rico-Fontalvo JE, Pájaro-Galvis N, et al. DPP-4 inhibitors and diabetic kidney disease: a narrative review. Kidney Med. 2021;3(6):1065-1073.
CrossRef - Wang M, Li M, Xie Y. Systematic review: DPP-4 inhibitors and rheumatoid arthritis risk. Endocr J. 2021;68(6):729-738.
CrossRef - Razavi M, Wei YY, Rao XQ, Zhong JX. DPP-4 inhibitors and GLP-1 receptor agonists: cardiovascular safety. Mil Med Res. 2022;9(1):45.
CrossRef - Epelde F. Transforming diabetes care: the expanding role of DPP-4 inhibitors. Medicina (Kaunas). 2024;60(11):1793.
CrossRef - Nasr NE, Sadek KM. Mechanisms of incretin-dependent therapies for diabetes. Environ Sci Pollut Res. 2022;29(13):18408-18422.
CrossRef - Chen YC, Chen TH, Sun CC, et al. DPP-4 inhibitors and autoimmune disease risk in type 2 diabetes: a nationwide cohort study. Acta Diabetol. 2020;57(10):1181-1192.
CrossRef - Singhal S, Patil VM, Verma S, Masand N. Recent advances and structure-activity relationship studies of DPP-4 inhibitors as anti-diabetic agents. Bioorg Chem. 2024;146:107277.
CrossRef - Liu Y, Zhao W, Jiang Y, Xing S, Li W. Study on the mechanism of interaction between dipeptidyl peptidase 4 and inhibitory peptides based on Gaussian accelerated molecular dynamic simulation. Int J Mol Sci. 2024;25(2):839.
CrossRef - Nada H, Calvo-Barreiro L, Cho S, et al. HTS-Oracle: A retrainable AI platform for high-confidence hit identification across difficult-to-drug targets. bioRxiv. Published online July 25, 2025.
CrossRef - Roach P. Transforming drug discovery through the fusion of AI-driven analysis and protein micropatterning. Expert Opin Drug Discov. 2025;20(12):1505-1511.
CrossRef - Saini K, Sharma S, Khan Y. DPP-4 inhibitors for treating T2DM-hype or hope? An analysis based on the current literature. Front Mol Biosci. 2023;10:1130625.
CrossRef - Vasquez-Martínez N, Trapala J, Álvarez-Añorve LI, et al. GPU-accelerated virtual screening and molecular dynamics simulations for identification of novel DPP-4 inhibitors. ACS Omega. 2026.
CrossRef - Al-Hamashi AA, Abdulhadi SL, Ali RM. Evaluation of zinc chelation ability for non-hydroxamic organic moieties. Egypt J Chem. 2023;66(5):215-221.
- Dav R, Imran M, Dhamija P, Chaurasia DK, Handu S. Virtual screening, ADMET prediction and dynamics simulation of potential compounds targeting the main protease of SARS-CoV-2. J Biomol Struct Dyn. 2020;39(17):6617-6632.
CrossRef - Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449-461.
CrossRef - Desmond molecular dynamics system. D E Shaw Research;2021. Maestro-Desmond interoperability tools. Schrödinger;2025.
- Srivastava S, Haneef M, Saxena VL, Khan M, Khan S. Structure and ligand-based in silico studies towards the natural inhibitors against receptor recognition spike protein of SARS-CoV-2. Open Bioinform J. 2024;17:e18750362284177.
CrossRef - Yahya M, Al-Hamashi A. Identification of selisistat derivatives as SIRT1-3 inhibitors by in silico virtual screening. Turk Comput Theor Chem. 2024;8(2):1-11.
CrossRef




