Pharmacology of Protein Binding of Drugs – An Extensive Review
 , Fareha Anan Shristy2, Tashfiya Zaman Mouree3, Saif Bin Salam Bondhon4 , Shaila Kabir5and Fahima Aktar5
, Fareha Anan Shristy2, Tashfiya Zaman Mouree3, Saif Bin Salam Bondhon4 , Shaila Kabir5and Fahima Aktar5 1Department of Pharmacy, School of Pharmacy and Public Health, Independent University, Bangladesh.
2Department of Pharmacy, Faculty of Pharmacy, University of Dhaka, Dhaka, Bangladesh.
3Department of Pharmacy, Atish Dipankar University of Science and Technology, Dhaka, Bangladesh.
4Department of Pharmacy, Bangladesh University of Professionals (BUP), Dhaka, Bangladesh.
5Molecular Pharmacology and Herbal Drug Research Laboratory, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Dhaka, Dhaka, Bangladesh.
Corresponding Author E-mail: amranmsspph@iub.edu.bd
DOI : http://dx.doi.org/10.13005/bpj/3419
Download this article as:
![]()
Protein binding is a fundamental determinant of drug pharmacokinetics and pharmacodynamics, influencing drug distribution, efficacy, toxicity, metabolism, and elimination. Most drugs interact with plasma proteins such as albumin, α1-acid glycoprotein (AAG), and lipoproteins, with the extent and nature of binding depending on physicochemical properties of the drug as well as physiological and pathological factors. Albumin predominantly binds acidic and neutral drugs, whereas AAG preferentially binds basic compounds. Lipoproteins serve as important carriers for highly lipophilic drugs, thereby affecting their distribution and clearance. The pharmacologically active fraction of a drug is the unbound portion, making protein binding a critical factor in determining therapeutic response and adverse effects. This review provides a comprehensive overview of the mechanisms and principles of drug-protein interactions, the major plasma proteins involved, and their influence on drug disposition. It further examines factors that alter protein binding, including disease states, age-related physiological changes, protein concentration variability, species differences, drug-drug interactions, food–drug interactions, and drug-metal interactions. The clinical implications of altered protein binding are discussed with reference to selected therapeutic agents, highlighting the importance of monitoring unbound drug concentrations in specific patient populations. By integrating classical concepts with recent advances in the understanding of plasma protein binding, this review underscores the pivotal role of drug–protein interactions in optimizing pharmacotherapy, improving therapeutic outcomes, and minimizing toxicity.
KEYWORDS:Albumin; α1-Acid glycoprotein; Drug disposition; Drug interactions; Drug-protein binding; Lipoproteins; Therapeutic drug monitoring
Introduction
Drug protein binding is a common feature in pharmacology and has been studied by researchers from many scientific areas. A drug’s behavior in the body largely depends on how it interacts with biologically active macromolecules. Because protein binding plays an important role in pharmacokinetics and drug protein interactions are relatively easy to study, a large amount of research has been published on this topic. Although much of this work is descriptive, important progress has been made in explaining, in physiological and quantitative terms, how protein binding affects drug distribution and therapeutic response. 1
Most drugs bind to proteins in plasma and tissues, but the extent of binding differs between drugs. Even for the same drug, protein binding may change due to disease, age, or the use of other medicines. Only the unbound or free fraction of a drug can cross biological membranes. This free drug can reach metabolic and transport sites and bind to its target to produce an effect. Therefore, understanding changes in protein binding is essential for explaining both drug’s movement in the body and drug action.2-3
The way a drug spreads through the body and is ultimately removed depends on several factors. First, the drug’s physical and chemical properties determine how easily it can reach binding sites outside the blood vessels. Second, the drug’s binding to plasma proteins affects its movement. This binding depends on how strongly the drug attaches to the protein, measured by the affinity constant (Kp). Third, the drug can also bind to proteins in tissues. This tissue binding is controlled by how many binding sites are available and how strongly the drug binds to them.4-5
Plasma protein binding influences drug distribution, action, and elimination. Drugs bound to albumin are transported in the bloodstream but cannot readily leave capillaries; therefore, tissue distribution depends mainly on the free, uncharged fraction in plasma. This free fraction is responsible for both therapeutic and toxic effects and determines the rate of drug elimination. Consequently, protein binding affects both the intensity and duration of drug action.
Protein binding plays a fundamental role in biological systems and drug discovery, encompassing interactions between proteins and other proteins, small molecules, oligonucleotides, and therapeutic compounds. Protein-protein binding affinity is determined by the change in free energy that occurs upon complex formation, where a greater decrease in free energy reflects stronger binding interactions.6 Similarly, protein-ligand interactions are essential for numerous cellular processes, including signal transduction, intracellular communication, and immune regulation, while also serving as a critical foundation for modern drug development. Advances in analytical techniques, particularly native mass spectrometry, have significantly improved the characterization of protein-ligand complexes by enabling sensitive and accurate assessment of binding interactions and stoichiometry.7 Beyond small molecules, protein interactions with oligonucleotides are increasingly important in therapeutic applications, where the chemical and structural determinants of binding influence efficacy and specificity.8 In pharmacology, plasma protein binding is a key determinant of drug disposition, influencing both pharmacokinetic and pharmacodynamic properties by regulating the balance between free and bound drug concentrations, thereby affecting efficacy and safety.9 Alterations in protein binding may occur under various physiological and pathological conditions, including hepatic and renal impairment, necessitating individualized dose adjustments to ensure optimal therapeutic outcomes.10 Furthermore, plasma protein binding is now recognized as a complex and dynamic process that can be strategically optimized during drug design to improve in vivo efficacy and clinical performance.11-12 Given the central importance of protein interactions in biological function and therapeutic development, accurately predicting ligand–protein binding affinities has become a crucial step in drug discovery, with machine learning approaches such as Graph Convolutional Networks demonstrating considerable potential for accelerating the identification and optimization of promising drug candidates.13
Most drugs bind to plasma proteins to some extent. Albumin primarily binds acidic and neutral drugs, whereas basic drugs bind less to albumin and more often to globulins. Strong protein binding can restrict drug movement into tissues, like the protective binding of bilirubin in newborns. Protein binding becomes clinically relevant when more than about 70% of a drug is bound, and for some drugs the free fraction is extremely small. Because binding is rapid and reversible, it usually does not limit drug uptake into well-perfused organs.14-15
Although plasma protein binding has been extensively studied for decades, recent advances have highlighted its broader clinical relevance in precision medicine, therapeutic drug monitoring, drug-drug interactions, and altered pharmacokinetics in special populations. However, many existing reviews focus primarily on classical pharmacokinetic principles and provide limited discussion of emerging topics such as food-drug interactions, drug-metal interactions, disease-associated alterations in protein binding, and translational implications for modern pharmacotherapy.15 Therefore, a comprehensive and updated review is warranted.
Materials and Methods
The total skeleton of the work was constructed using modified PRISMA method.16 A comprehensive literature search was conducted using databases and search platforms such as PubMed, ResearchGate, Google Scholar, and additional search engines, including artificial intelligence-assisted tools. The search strategy employed terms such as “drug-protein binding”, “plasma proteins”, “albumin”, “α1-acid glycoprotein”, “Drug disposition” and “lipoproteins”, “Drug interactions”, “Therapeutic drug monitoring” among others, to locate pertinent studies and gather information across different aspects of the topic. Relevant articles from our prior publications were also reviewed to strengthen and expand the discussion.17-31
Findings and Discussion
Drug-Protein Binding: Theoretical Framework
Drug binding to plasma proteins is generally treated as a fast and reversible equilibrium process that follows the law of mass action. However, some drugs have been shown to bind irreversibly to plasma proteins under certain conditions.32
In the simplest scenario, a protein possesses a single reversible binding site for a drug molecule. The interaction between the drug (D) and the protein (P) can be represented by the equilibrium:
D + P ⇌ DP
Here, [D], [P], and [DP] denote the molar concentrations of unbound drug, unbound protein, and the drug–protein complex, respectively. The forward and reverse reactions are governed by the association rate constant (kon) and dissociation rate constant (k0ff).
At equilibrium, the rate at which the drug binds to the protein equals the rate at which it dissociates. This balance allows the association constant (Ka ) to be defined as:
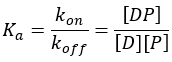
The dissociation constant (Kd), which is the inverse of Ka, is also commonly reported. Kd corresponds to the concentration of free drug required to occupy half of the available binding sites at equilibrium.
Another key parameter is the extent of binding, which can be expressed as the number of drug molecules bound per protein molecule, or the fraction of occupied binding sites. This parameter is represented by r:
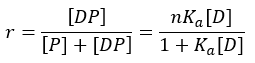
In this equation, ‘n’ represents the maximum number of binding sites available on each protein molecule. It is assumed that all ‘n’ sites have identical affinity for the drug. A protein may contain several different classes of binding sites, each characterized by its own affinity constant. When multiple binding-site classes exist, the expression for r becomes:
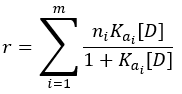
Here, ‘m’ represents the number of distinct binding-site classes, each with its own number of sites ni and association constant Kai.
This model assumes that each binding site interacts with the drug independently, which is valid only when there are no allosteric effects. If binding at one site alters the affinity at another, more complex models are required. Additionally, these equations assume that binding at each site is a single-step, reversible process. When binding involves multiple steps or irreversible mechanisms, the simple equilibrium models no longer applies.33
Methods for Determining Drug-Protein Binding Parameters
Direct Method
In this approach, the binding ratio r (amount of drug bound per unit protein) is plotted directly against the free drug concentration [D] according to the binding equation. The resulting graph forms a hyperbolic curve, like the curve observed in enzyme kinetics.
Important characteristics of this curve include:
- At the plateau, the value of r equals n, which represents the total number of binding sites.
- When r = n/2, the corresponding drug concentration equals 1/Ka, where Ka is the association constant.
- The initial slope near the origin equals Ka.
This technique reduces errors caused by transformation of variables. However, the hyperbolic shape makes accurate graphical interpretation difficult. Therefore, the parameters are usually estimated using computer-based nonlinear regression.
If two types of binding sites exist, the curve does not reach a simple asymptote and instead shows a change in slope. The initial portion represents high-affinity sites, while the later section reflects the combined contribution of both site classes.
Lineweaver–Burk Method (1934)
This method linearizes the binding equation by taking its reciprocal form. When the inverse values 1/r are plotted against 1/[D], a straight line is obtained.
From this linear plot:
- The intercept on the y-axis equals 1/n, where n is the number of binding sites.
- The slope of the line equals 1/(nKa).
Because the relationship becomes linear, the data can be analyzed easily using simple linear regression. However, this method has an important limitation: the transformation exaggerates errors, especially when r values are small. Consequently, the method lacks precision and is generally suitable only when the drug interacts with a single class of binding sites.
Scatchard Method (1949)
The Scatchard method rearranges the binding equation into the form:
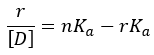
In this analysis, r/[D] is plotted against r. The graph produces a straight line with the following properties:
- Slope = −Ka
- Intercept on the r-axis = n
- Intercept on the r/[D] axis = nKa
This graphical method allows simultaneous estimation of the binding affinity (Ka) and the number of binding sites (n).
If only one class of independent binding sites exists, the plot is linear. However, when multiple binding site types are present, the plot becomes curved, indicating heterogeneity in binding affinity.34 A schematic diagram of all methods for determination of drug-protein interaction is shown in Figure 1.34
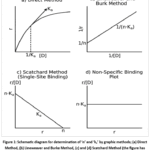 |
Figure 1: Schematic diagram for determination of ‘n’ and ‘ka’ by graphic methods; (a) Direct Method, (b) Lineweaver and Burke Method, (c) and (d) Scatchard Method. |
Protein Binding and Drug Disposition
Distribution
At equilibrium, drug concentration in extracellular fluids equals the unbound drug concentration in plasma. Cerebrospinal fluid (CSF), which contains minimal albumin, generally mirrors the free drug level in plasma. The apparent volume of distribution (Vd) indicates how widely a drug distributes and how extensively it binds. Highly protein-bound drugs show low Vd values when calculated from total plasma concentrations (e.g., phenylbutazone ~0.1 L/kg). In contrast, drugs with extensive tissue binding have large Vd values, such as digoxin (~6 L/kg) and chlorpromazine (~20 L/kg).
Renal Excretion
Only the unbound fraction of a drug can be filtered at the glomerulus; protein-bound drug is not filtered. Therefore, protein binding can prolong the half-life of drugs that are neither actively secreted nor rapidly metabolized. When a drug is actively secreted in the renal tubules, protein binding generally does not limit excretion and may even enhance it by keeping the drug in circulation for delivery to the kidneys.
Hepatic Elimination
Although long-acting sulfonamides historically suggested that high protein binding alone causes prolonged half-life, many highly bound drugs are rapidly metabolized and eliminated. Drug half-life varies across species, emphasizing that hepatic and renal elimination efficiency is more decisive than protein binding. When hepatic uptake and metabolism are efficient, protein binding mainly functions as a transport reservoir.
Flow-dependent elimination: For drugs such as propranolol, metabolic rate depends on blood flow to the liver, since delivery limits elimination.
Flow-independent elimination: For drugs like warfarin, only the unbound fraction is extracted by the liver; thus, protein binding acts mainly as a storage reservoir.14
Figure 2 illustrates protein binding and drug distribution in our physiological system.14
Figure 2: Protein binding and drug distribution in human body (the figure has been modified from reference 14).
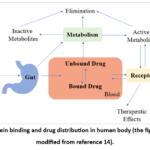 |
Figure 2: Protein binding and drug distribution in human body (the figure has been modified from reference 14). |
Plasma Binding Proteins
Human plasma contains numerous potential drug-binding components, but the most significant contributors are human serum albumin (HSA) and α1-acid glycoprotein (AAG), which are abundant and can bind many drugs with enough strength to meaningfully influence their distribution and pharmacological effects. Other plasma constituents, such as globulins and lipoproteins, may also bind drugs, but their impact is generally smaller.33
Albumin: Albumin is the main protein in plasma, and its concentration remains relatively constant under normal physiological conditions, varying by less than two-fold. It is a key contributor to the plasma protein binding of many basic drugs, often accounting for a large portion of their total binding. For example, about half of the propranolol present in plasma is bound to albumin.35 Albumin is also the primary plasma and tissue protein involved in nonspecific drug binding. The drug-albumin binding and the structure of albumin are shown in Figure 3 and Figure 4.
Many acidic drugs are strongly bound to plasma albumin, which significantly limits their distribution within the body. Phenytoin illustrates this effect, with an apparent volume of distribution of about 0.64 L/kg, indicating that only the unbound fraction is able to reach the brain.
Although albumin generally shows low-affinity, high-capacity binding for basic drugs, meaning changes in its concentration usually have little effect on their protein binding, marked alterations in albumin levels can still affect drug binding. Hyperalbuminemia is rare, but hypoalbuminemia may occur in severe liver or renal disease, particularly in nephrotic syndrome. Chronic ambulatory peritoneal dialysis (CAPD) for chronic renal failure can also cause substantial albumin loss, leading to hypoalbuminemia of a severity comparable to nephrotic syndrome. These changes may alter the plasma protein binding of some basic drugs, depending on how much albumin contributes to their total binding.36-38
The skin contains the largest proportion of extravascular albumin, representing about 18% of the total exchangeable albumin in the body. A fluorescent probe displacement technique was used to identify two distinct drug-binding regions on albumin as classified in Table 1: site I (the warfarin-binding site) and site II (the benzodiazepine-binding site).39
Table 1: Classification of common drugs by human serum albumin binding site.
| Binding Site | Drugs’ name |
| Site I | Warfarin, Phenylbutazone, Azapropazone, Indomethacin, Tolbutamide, Iodipamide, Iophenoxic acid, Furosemide, Bucolome, Sulfisoxazole. |
| Site II | Diazepam, Diflunisal, Ibuprofen, Ketoprofen, Naproxen, 6-MNA, Diclofenac, Etodolac, Clofibrate, Iopanoic acid. |
Here Figure 3 shows Reversible binding of a drug to Albumin and retention of protein-bound drug within vasculature and Figure 4 shows the structure of albumin.
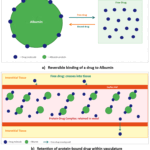 |
Figure 3: Drug-albumin binding. |
 |
Figure 4: Structure of Serum Albumin. |
Alpha1-acid glycoprotein (AAG)
AAG is found in plasma at concentrations that are typically about 100 times lower than those of albumin. Its physiological function is not well understood, although it may be involved in normal coagulation, immune responses, and tissue repair processes. AAG is a glycoprotein with a molecular weight of approximately 40,000. Its high sialic acid content gives it an acidic character and a low pKa. Drug binding to AAG appears to be driven more by hydrophobic interactions than by electrostatic forces, since removal of sialic acid residues does not significantly decrease binding.40
Certain acidic drugs, such as warfarin, can compete with basic drugs for what seems to be a single binding site, possibly located on the protein portion of the glycoprotein. These acidic drugs are mainly those that bind at site I (for example, warfarin), rather than site II drugs like phenylbutazone. It is unlikely that AAG contributes substantially to the binding of these acidic drugs in plasma because albumin’s affinity and binding capacity for them is so high.
Pharmacokinetic studies using radiolabeled AAG show that about 60% of the protein resides in the central compartment (likely plasma), while the remaining portion is found in a peripheral compartment, probably the extravascular space. It is uncommon for basic drugs to bind exclusively to AAG, although disopyramide and erythromycin may do so at therapeutic levels.41 However, due to the specific binding properties of AAG, it frequently represents the primary factor driving variability in plasma protein binding both between different individuals and within the same individual over time.
Variability in the plasma protein binding of basic drugs is partly attributable to significant fluctuations in AAG concentrations in both healthy and diseased states. Additionally, AAG acts as a high-affinity, low-capacity binding site, meaning it can be readily saturated as drug concentrations increase. Table 2 lists several drugs for which AAG is a major determinant of plasma protein binding.36
Table 2: Examples of basic drugs with significant binding to α₁-acid glycoprotein (AAG).
| Drug Category | Drugs |
| Antiarrhythmics | Aprindine, Bupivacaine, Disopyramide, Lignocaine, Pirmenol, Quinidine, Verapamil. |
| Antidepressants | Amitriptyline, Imipramine, Nortriptyline. |
| β-adrenoceptor blockers | Alprenolol, Oxprenolol, Pindolol, Propranolol, Timolol. |
| Miscellaneous | Chlorpromazine, Dipyridamole, Erythromycin, Metoclopramide, Nicardipine, Phencyclidine, Prednisolone, Progesterone, Triazolam. |
| Opiates | Methadone, Pethidine. |
Lipoproteins
Lipoproteins are lipid–protein complexes that occur as spherical or disc-shaped particles, with sizes spanning from approximately 35 Å to as large as 6 μm in diameter. Owing to their heterogeneous composition and particle dimensions, lipoproteins exhibit physicochemical behavior comparable to emulsified systems. High-density and low-density lipoproteins (HDL and LDL) resemble microemulsions, whereas very-low-density lipoproteins (VLDL) and chylomicrons behave more like macroemulsions. Structurally, lipoproteins possess a hydrophobic core composed predominantly of neutral lipids, including triacylglycerols and cholesteryl esters. This core is encased by an outer layer formed by a single phospholipid monolayer, which confers aqueous solubility through its polar head groups. Embedded within this surface layer are amphipathic constituents such as unesterified cholesterol and specialized transport proteins known as apolipoproteins.
Lipoproteins are commonly categorized according to two distinct classification schemes: particle density and electrophoretic behavior. Based on density, lipoproteins are designated as chylomicrons (δ < 0.94 g/mL), VLDL (0.94 < δ < 1.006 g/mL), LDL (1.006 < δ < 1.063 g/mL), and HDL (1.063 < δ < 1.21 g/mL). Alternatively, separation by electrophoretic mobility classifies HDL as α-lipoproteins, VLDL as pre-β-lipoproteins, and LDL as β-lipoproteins. Chylomicrons, due to their large size and low density, do not exhibit measurable electrophoretic migration.Top of Form
Drug–lipoprotein interactions differ fundamentally from drug binding to plasma proteins such as albumin, α₁-acid glycoprotein, or steroid-binding globulins, primarily due to the distinctive structural and compositional characteristics of lipoproteins. Unlike soluble proteins, lipoproteins provide multiple interaction environments, allowing drugs to associate either with the hydrophobic lipid core or with the more polar components at the particle surface.
Compounds that associate with lipoproteins are typically highly lipophilic and frequently undergo extensive metabolism, which often represents a major route of their elimination from the body. Several drugs with elevated log P values have been shown to exhibit substantial affinity for lipoproteins, including cyclosporine A, amiodarone, halofantrine, amphotericin B, nystatin, and eritoran.42-43 Table 3 summarizes representative drugs whose interactions with lipoproteins have been studied in detail. The interaction is categorized into four main carriers: TRL (Triglyceride-rich lipoproteins), LDL (Low-density lipoproteins), HDL (High-density lipoproteins), and LPDP (Lipoprotein-deficient plasma fraction).42
Table 3: Key Drug-Lipoprotein Interactions.
| Drugs | Primary Association | Pharmacological Outcome |
| Amphotericin B | LDL, LPDP | Linked to significant kidney toxicity. |
| Abelcet | TRL, HDL | Demonstrates reduced kidney toxicity. |
| Cyclosporine A | TRL, LDL, HDL | Linked to significant kidney toxicity. |
| Halofantrine | TRL, LDL, HDL | Results in enhanced drug activity. |
| Clozapine | HDL, LPDP | Results in enhanced drug activity. |
| Haldol | HDL, LPDP | Results in enhanced drug activity. |
| Eritoran | TRL, HDL | Drug de-activation occurs specifically via HDL. |
| Amiodarone | All (TRL, LDL, HDL, LPDP) | Leads to a potential increase in drug activity. |
| Paclitaxel | HDL, LPDP | Shows a decrease in toxic side effects. |
Below Figure 5 shows the structure of Lipoprotein and Globulin
 |
Figure 5: Lipoprotein (a) and Globulin (b) Structure. |
Influence of Plasma Protein Binding on the Pharmacokinetic Behavior of Selected Drugs
Propranolol
Plasma and hepatic protein binding significantly influence the disposition of propranolol across species. The free drug concentration determines the apparent volume of distribution (Vd), such that increased free propranolol raises Vd without altering the volume of distribution of unbound drug. Since clearance remains constant, reduced Vd leads to faster elimination, illustrating that plasma binding can directly affect half-life even for drugs cleared solely by metabolism. Across species (monkey, dog, man, rat), Vd correlates with free drug concentration despite large differences in clearance. Additionally, perfused rat liver studies revealed a saturable, high-affinity hepatic binding site, contributing to nonlinear, dose-dependent elimination kinetics.44
Phenytoin
Phenytoin is extensively bound to plasma proteins, primarily albumin (85–95% at therapeutic levels), with smaller binding to α-globulins. Protein binding significantly influences its distribution, as unbound phenytoin correlates closely with concentrations in CSF, erythrocytes, and saliva. Because total plasma concentrations may not reflect pharmacologic effect, unbound levels provide a better distinction between toxic and non-toxic patients. Binding decreases in conditions such as uremia, hyperbilirubinemia, fetal serum, and hypoalbuminemia, leading to lower total plasma concentrations, increased volume of distribution, and higher risk of adverse effects. Displacement by drugs such as salicylates and phenylbutazone further reduces binding. Due to these factors, monitoring unbound phenytoin is more clinically reliable than total plasma concentration.45
Corticosteroid
Corticosteroid effects are determined by their unbound plasma fraction, as free steroid concentrations govern biological activity and adrenal feedback. Steroids bind both albumin and high-affinity globulins such as corticosteroid-binding globulin (CBG/transcortin) and gonadal binding globulin (GBG). CBG and GBG have high affinity but low plasma concentrations (~10⁻⁸ M), so they saturate at elevated steroid levels, whereas albumin, despite lower affinity, binds more steroid due to its higher concentration (~10⁻⁴ M). Consequently, steroid distribution and clearance differ between physiological and pharmacological doses; for example, cortisol’s volume of distribution increases from ~10–17 L at normal doses to ~70–80 L at high doses. Pregnancy increases both free cortisol exposure and CBG levels, which helps prevent cortisol excess symptoms. Overall, protein binding significantly influences steroid pharmacokinetics and pharmacodynamics.46
Anticoagulants
Coumarin anticoagulants are highly protein-bound, and binding differences strongly influence their distribution and elimination. Warfarin clearance is linearly related to the unbound fraction (0.002–0.015), supporting a pharmacokinetic model where clearance varies with the free fraction. In hypoalbuminemia, warfarin elimination is accelerated, as demonstrated by a patient whose half-life shortened from ~2 days to ~6 hours, consistent with increased free drug availability at low albumin levels. Similar concentration-dependent binding effects have been observed for dicoumarol, where variability in liver-to-serum ratios correlates with half-life, indicating that protein binding governs hepatic distribution and biotransformation.47
Antibiotics
Sulfonamide dosing depends strongly on protein binding because free drug concentration governs both therapeutic effect and elimination. Maintenance doses varied 13-fold among sulfonamides despite only a 4-fold difference in antimicrobial activity, indicating protein binding is a major determinant of dosing requirements. Protein binding also influences metabolism, as higher free and nonionized fractions correlate with faster acetylation rates. For penicillin derivatives, binding both slows elimination and reduces active free drug, affecting overall drug exposure in opposite directions.48
Bilirubin
Strong albumin binding can increase bilirubin toxicity risk. Premature infants treated with highly albumin-bound sulfonamides had a higher incidence of bilirubin encephalopathy, suggesting drug-induced displacement of bilirubin. Bilirubin binds to high-affinity and secondary albumin sites; anionic drugs can displace bilirubin from the secondary sites, increasing free bilirubin levels. Newborn plasma binds bilirubin less strongly than adult plasma, contributing to higher susceptibility to toxicity. Phenobarbital may reduce bilirubin by inducing liver ligandin, enhancing bilirubin uptake and storage.49
Other Drugs
Naproxen renal clearance increases disproportionately at high plasma concentrations but becomes linear with dose when corrected for protein binding, indicating binding affects its elimination. Imipramine shows wide interpatient variability in protein binding, making unbound concentrations a better predictor of response than total plasma levels. In radio-opaque agents, higher protein binding favors biliary excretion, while lower binding favors renal excretion, and binding strength correlates with intravenous toxicity across species.50
Factors That Alter Drug-Protein Binding
Effect of Diseases
Drug disposition is highly influenced by physiological and pathological alterations. Variations in plasma protein binding affected by drug concentration, protein affinity, or disease states alter free drug levels, impacting therapeutic efficacy and adverse drug reaction risk. Key conditions include hepatic dysfunction, renal failure, inflammation, and malnutrition.
Alterations in drug-albumin binding arise from reduced albumin concentrations, conformational modifications, and/or competitive displacement by endogenous substances. Drugs with high protein binding (>90%) are especially sensitive: even a reduction from 99% to 98% binding doubles the free fraction. Monitoring free rather than total drug concentrations provide a more accurate assessment of pharmacologically active levels in affected patients. 51
Hypoalbuminemia (serum albumin <35 g/L), seen in chronic liver disease, malnutrition, burns, nephrotic syndrome, and severe infections, increases free drug fractions of highly bound drugs. Phenytoin and valproic acid (>90% albumin-bound) can cause neurotoxicity despite apparently normal total plasma levels, while warfarin (~99% bound) may produce elevated bleeding risk, both highlighting the need for free drug monitoring and dose adjustment.52
Renal Impairment
In nephrotic syndrome, albumin drops to 7-25 g/L (normal ~42 g/L), with free fractions of phenytoin and clofibrate increasing by ~90% and ~200%, respectively. In chronic renal failure and uremia, reduced binding extends beyond hypoalbuminemia to include accumulation of uremic toxins (indoxyl sulfate, hippuric acid, CMPF) competing at albumin binding sites I and II, carbamylation of albumin, and oxidative modifications reducing site II capacity. Consequently, drugs such as furosemide, diazepam, and phenytoin exhibit altered pharmacokinetics, with increased free concentrations causing enhanced effects or toxicity despite normal total drug levels.52-53
Liver Disease
Decreased albumin (~30 g/L) in hepatic disorders results mainly from altered degradation and redistribution rather than reduced synthesis. Elevated bilirubin, more than albumin levels correlates with increased free phenytoin in hepatitis and cirrhosis. Cirrhotic serum specifically inhibits site II (diazepam-binding site) binding, prompting the clinical use of albumin binding capacity at site II (ABiC) as a marker of liver failure severity and a monitoring tool for liver support interventions. Other conditions like diabetes, shock, trauma, and cancer similarly affect albumin binding via concentration changes, structural modifications, or endogenous displacement. 39,53
Inflammation and α1-Acid Glycoprotein (AAG)
Unlike albumin, AAG is an acute-phase protein that rises during inflammation, trauma, cancer, and myocardial infarction. Elevated AAG increases binding of basic drugs (e.g., lidocaine, propranolol, imipramine, docetaxel), reducing their free fraction and potentially necessitating higher doses. Inflammatory cytokines (IL-6, TNF-α) also downregulate CYP enzymes, raising substrate drug concentrations. Additionally, inflammation-associated downregulation of L-type calcium channel proteins reduces verapamil’s effectiveness while promoting its accumulation and toxicity. 54-55
Malnutrition
Reduced albumin synthesis in protein-energy malnutrition increases free fractions of highly bound drugs (diazepam, prednisolone, salicylates), risking exaggerated pharmacological responses. This is especially relevant in pediatric and critically ill patients with severe nutritional deficiencies, where careful dosing and therapeutic monitoring are essential. 56
Age
Multiple investigations have demonstrated that plasma protein binding of drugs is substantially reduced during fetal life and the neonatal period when compared with adults. Drugs such as ampicillin, benzylpenicillin, phenobarbital, and diphenylhydantoin exhibit the highest degree of protein binding in adults, intermediate binding in neonates, and the lowest binding in fetuses. This age-related pattern is partly explained by the reduced plasma albumin concentrations present before and shortly after birth. Comparable reductions in binding have also been reported for several other compounds, including imipramine, diazoxide, bupivacaine, lidocaine, sulfaphenazole, salicylates, and bilirubin.57
In addition to quantitative differences in albumin levels, qualitative differences in albumin binding capacity appear to contribute to diminished drug binding in early life. Sulfaphenazole binds with lower affinity to albumin derived from umbilical cord and neonatal plasma than to adult albumin. Notably, when cord plasma was treated with activated charcoal, the binding affinity of sulfaphenazole increased to values comparable to those observed in adults. This finding suggests that the reduced binding capacity of neonatal albumin results from competition with endogenous ligands, such as bilirubin, that occupy albumin binding sites and can be removed by adsorption.58
Age-dependent changes in drug binding have also been described for plasma proteins other than albumin. For example, the corticosteroid-binding protein transcortin exhibits markedly reduced binding capacity in umbilical cord plasma approximately 50% of that observed in healthy adult males. This reduced capacity persists throughout the first month of life but gradually increases thereafter, reaching adult levels by approximately one year of age. The maturation of transcortin binding capacity parallels the age-related increase in circulating transcortin concentrations.1
Consistent with these observations, clinical studies have identified significant correlations between age and the fraction of unbound drug in plasma. Mather et al.59 reported a positive association between increasing age and the unbound fraction of meperidine, while Hadjian et al.60 demonstrated similar age-related effects for cortisol binding to transcortin. Collectively, these findings highlight the profound influence of developmental physiology on plasma protein binding and underscore the importance of considering age-dependent changes when evaluating drug disposition in fetuses, neonates, and infants.1
Protein Concentration
Disease states and physiological disturbances frequently alter plasma and tissue protein concentrations, leading to complex, nonlinear changes in drug–protein binding governed by mass-action principles. Drugs with low affinity show binding roughly proportional to protein concentration, whereas highly bound drugs remain largely unaffected by protein changes except at very low protein levels; compounds with intermediate affinity depend on both drug and protein concentrations. These relationships indicate that percent binding alone is insufficient for interpretation unless drug and protein concentrations are reported, and binding parameters cannot always be extrapolated across protein concentrations, as demonstrated for thiopental and other agents.
Clinically, reduced albumin levels are associated with increased adverse drug reactions and altered dosage requirements. Large surveillance studies have shown higher rates of side effects with prednisone and diphenylhydantoin in patients with hypoalbuminemia. Similarly, altered globulin and albumin concentrations in hepatic disease influence the dose requirements of neuromuscular blocking agents such as tubocurarine and alcuronium.61
Changes in protein binding also affect drug elimination and diagnostic testing. In liver cirrhosis, decreased albumin and increased bilirubin correlate with prolonged chloramphenicol half-life, reflecting impaired hepatic metabolism. Hypoalbuminemia can mask hepatic dysfunction in sulfobromophthalein testing, resulting in false-negative outcomes. Protein concentration further influences the plasma handling of endogenous compounds, including calcium and steroid hormones, necessitating protein-corrected clinical interpretation.62
In addition to quantitative changes, qualitative alterations in albumin structure occur in disease states such as rheumatoid arthritis, leading to modified drug binding independent of protein concentration. Together, these findings emphasize that disease-related changes in plasma proteins significantly affect drug binding, disposition, and clinical response.1
Species Differences in Binding
Plasma protein binding of both acidic and basic drugs varies markedly among species. Such interspecies differences have been documented for numerous compounds, including salicylic acid, penicillins, sulfonamides, phenylbutazone, diphenylhydantoin, amphetamines, propranolol, chlorpromazine, and rifampicin. These variations arise partly from species-dependent differences in plasma protein concentrations, but more importantly from genetic differences in albumin structure, which influence binding affinity and the number of available binding sites for a given drug.63
The magnitude of these differences can be substantial. For example, salicylic acid binding is relatively high in humans and certain laboratory animals (approximately 50–90%, depending on concentration), but markedly lower (<20%) in species such as rats and dogs. Such variability in protein binding contributes significantly to interspecies differences in pharmacokinetics, pharmacodynamics, and toxicity, and represents a major limitation in extrapolating animal data to humans.
Clear examples of this effect have been demonstrated for chlorpromazine, which shows minimal protein binding in rats, intermediate binding in dogs, and the highest binding in humans, resulting in corresponding species differences in volume of distribution and drug disposition rates. Comparable interspecies trends have also been observed for propranolol.44
Antibiotics
Evidence suggests that antibodies formed against certain drugs can markedly increase their binding in serum. In patients hypersensitive to para-aminosalicylic acid, serum binding of the drug is significantly higher than in nonallergic individuals, indicating antibody involvement.64 Similarly, preferential binding of morphine to γ-globulins has been observed in sera from drug addicts, consistent with the formation of drug-specific antibodies following repeated antigenic exposure.
Although antibodies are well established as biological antagonists for macromolecules, toxins, and peptide hormones, their application against low–molecular-weight drugs have been limited. Nonetheless, compelling evidence exists for digoxin: administration of digoxin-specific antibodies protects animals from otherwise lethal cardiotoxic effects and profoundly alters drug disposition. Antibody binding produces markedly elevated serum digoxin concentrations, reduced renal excretion, and a dramatic prolongation of elimination half-life, while simultaneously reversing digoxin-induced arrhythmias by removing the drug from myocardial tissue.65
Drug Interactions
Plasma proteins have limited binding sites, so drugs may compete and displace each other, increasing the free drug fraction. This can enhance drug effect, tissue distribution, and elimination, while lowering total plasma concentration. Displacement depends on whether drugs bind the same site and their relative affinities and concentrations.
Clinically important examples include warfarin displacement by phenylbutazone and chloral hydrate metabolites, which increases bleeding risk and clearance. Other notable interactions are displacement of tolbutamide (hypoglycemia), methotrexate (toxicity), and tricyclic antidepressants by highly bound drugs. Bilirubin displacement by drugs such as sulfasoxazole can cause neonatal encephalopathy. Drug-induced albumin modification (e.g., aspirin acetylation) can also change binding for other drugs. Finally, urate displacement by uricosuric drugs and acidic compounds increases free urate and renal excretion, potentially triggering gout.66
Plasma Protein Binding and Its Impact on Drug Performance
Plasma protein binding is a key factor in how drugs behave inside the body. Since plasma proteins are present in much higher amounts than drug targets, only a small fraction of a drug remains free in the blood. This free drug is the part that can cross barriers, bind to receptors, and produce an effect. When a drug binds strongly to proteins, its apparent strength in the body is reduced. This idea is known as the Free Drug Principle and helps explain differences between lab results and effects seen in living systems.67
In most cases, drug activity follows this principle, even for drugs acting inside cells or in the brain. When it does not, it often suggests slow tissue entry, slow release from the target, or the involvement of active transport processes. Sometimes, these differences reveal limits in laboratory tests or suggest that a drug may act through another pathway.
Whole tissue drug levels, such as in the brain or tumors, should be interpreted carefully. High levels often reflect nonspecific binding rather than true drug action. When comparing data between species, differences in plasma protein binding must also be considered, as they can affect safety and dose estimates. During drug optimization, the balance between potency and free drug fraction is important. A drug with moderate potency but a higher free fraction may perform better in the body than a very potent drug that is mostly protein bound.15
Drug-Food Interaction
Food can influence how drugs bind to plasma proteins and may therefore affect their pharmacokinetics and pharmacological effects. Certain components of food, such as fatty acids, vitamins, and plant-derived polyphenols, may compete with drugs for binding sites on plasma proteins like albumin and α₁-acid glycoprotein.68-70 For example, after a high-fat meal, the concentration of free fatty acids in the blood may increase, and these fatty acids can compete with drugs for albumin binding sites. As a result, the fraction of free (unbound) drug in plasma may increase temporarily. Since only the free drug is pharmacologically active, such displacement may enhance drug distribution, therapeutic effects, or even toxicity in some cases. This effect has been observed with drugs such as diazepam and valproic acid, where increased fatty acid levels can reduce albumin binding and increase the unbound drug fraction.71
Food can also influence drug response through specific nutrient–drug interactions. A well-known example involves warfarin, a highly albumin-bound anticoagulant. Foods rich in vitamin K, such as spinach, broccoli, and other green leafy vegetables, can reduce the anticoagulant effect of warfarin by promoting the synthesis of clotting factors.72-73 Although this interaction mainly affects the drug’s pharmacodynamic action, variations in plasma protein binding may also contribute to changes in the free drug fraction. In addition, certain beverages can alter drug disposition. Grapefruit juice, for instance, can increase the systemic levels of several drugs including felodipine, cyclosporine, and simvastatin by inhibiting intestinal CYP3A4 enzymes. Such changes in metabolism may indirectly affect the balance between bound and unbound drug in circulation.74
Plant-derived compounds present in foods and beverages may also interact with plasma proteins. Flavonoids and other polyphenols found in tea, berries, and soy products have been shown to bind to albumin and may compete with some drugs for similar binding sites in vitro.75 Although most dietary effects on protein binding are relatively small, they may become clinically important for drugs that are highly protein bound and have a narrow therapeutic range. Therefore, dietary habits and nutritional status should be considered when evaluating drug therapy and interpreting variations in drug response. Figure 6 shows the overview of food drug interaction.
 |
Figure 6: Schematic diagram of Food-Drug Interaction. |
Drug-Metal Interaction
Drug–metal interactions should be considered, along with plasma protein binding, to provide a comprehensive understanding of drug disposition. Metal ions can interact directly with drugs, alter protein conformation, or compete for binding sites, ultimately modifying drug–protein affinity. These interactions are particularly relevant for metallodrugs, drugs capable of chelation, and conditions involving altered metal ion homeostasis (e.g., iron overload, copper imbalance, or therapeutic metal administration). Alterations in binding can change the free drug fraction, leading to either increased toxicity or reduced therapeutic efficacy.76,77
Endogenous or exogenous metal ions can significantly influence the binding of drugs to plasma proteins. Metal ions such as Ca²⁺, Zn²⁺, and Cu²⁺ can bind to albumin and induce conformational changes that alter drug-binding sites. These structural modifications may either enhance or reduce drug affinity for plasma proteins.
For instance, warfarin and ibuprofen, which bind to specific sites on albumin, may exhibit altered binding in the presence of metal ions that compete for or modify these sites. Metal-induced conformational changes in albumin have been shown to influence drug-binding capacity, potentially altering the free fraction of highly bound drugs.52,54
Certain drugs possess functional groups capable of chelating metal ions, forming drug–metal complexes that exhibit altered protein-binding characteristics. This is particularly relevant for tetracyclines and fluoroquinolones, which can chelate divalent and trivalent metal ions such as Ca²⁺, Mg²⁺, and Fe³⁺.
For example, ciprofloxacin forms complexes with metal ions that may reduce its free concentration as shown in the Figure 7 and alter its interaction with plasma proteins. These metal–drug complexes often display different binding affinities compared to the parent drug, thereby influencing drug distribution and bioavailability. In some cases, the formation of such complexes reduces protein binding by increasing hydrophilicity, while in others it enhances binding through altered molecular conformation.78
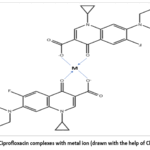 |
Figure 7: Ciprofloxacin complexes with metal ion (drawn with the help of ChemDraw). |
Metal-based drugs (metallodrugs) exhibit unique binding behaviors due to their ability to form coordination complexes with proteins. A classic example is cisplatin, a platinum-based anticancer drug, which binds extensively to plasma proteins including albumin and small proteins such as ubiquitin. Studies have demonstrated that cisplatin can bind to multiple sites on proteins, forming stable adducts that influence both drug distribution and toxicity.
Similarly, ruthenium-based anticancer agents such as KP1019 exhibit strong binding to albumin and transferrin in plasma. Albumin acts as a carrier, facilitating drug transport and possibly modulating cellular uptake. This extensive protein binding can prolong circulation time but may also reduce the free drug fraction available for pharmacological action.
Metal-binding plasma proteins such as transferrin and ceruloplasmin can also participate in drug–metal interactions. Transferrin, which primarily transports iron, has been shown to bind certain metal-based drugs and facilitate their transport in circulation. This mechanism is particularly important in the pharmacokinetics of ruthenium-based anticancer agents, which exploit transferrin pathways for targeted delivery to tumor cells.79
Additionally, competition between endogenous metals and drugs for binding to transferrin may influence drug disposition. For instance, in conditions of iron overload, altered transferrin saturation may affect the binding and transport of metallodrugs, potentially modifying their therapeutic efficacy.
Optimizing the dosage strategies, monitoring the free drug concentration and interactions of the metal ions are crucial to ensure the desired safety and therapeutic outcome among the patients receiving metal-based therapies and patients with compromised metal homeostasis.
Limitations and Future Perspectives
This review has several limitations. As it relies on published literature, the findings may be influenced by publication bias and variability in study designs, experimental methods, patient populations, and drug classes. Differences in protein-binding assessment techniques, including in vitro and in vivo approaches, may also contribute to inconsistencies across studies. Although foundational studies were included to provide historical context, some concepts may have evolved with recent advances in analytical and computational pharmacology. Furthermore, this review is qualitative in nature and does not include a systematic meta-analysis, limiting quantitative assessment of the available evidence. The clinical relevance of drug-protein interactions is further complicated by factors such as genetic variability, polypharmacy, nutritional status, and other patient-specific characteristics, which are not consistently addressed in the literature.
Recent advances in artificial intelligence (AI) and machine learning have substantially improved the prediction of protein–drug interactions and binding affinities, accelerating drug discovery. However, current models are primarily trained on experimental datasets that may not fully reflect the complex physiological conditions influencing protein–drug binding in vivo. Factors such as protein conformational flexibility, allosteric effects, and environmental influences can limit the accuracy of predictions based on static structural data. In addition, variability in data quality and the limited availability of high-quality binding datasets may affect model performance. Despite these challenges, AI-based approaches show considerable promise for identifying and optimizing therapeutic candidates. Future research should integrate dynamic protein behavior, standardized experimental data, and physiologically relevant models with advanced computational approaches to improve the prediction of drug disposition, therapeutic response, and personalized treatment outcomes.80
Conclusion
Protein binding remains a fundamental determinant of drug disposition and therapeutic response, influencing the pharmacokinetic and pharmacodynamic behavior of numerous medications. The interactions of drugs with plasma proteins such as albumin, α1-acid glycoprotein (AAG), and lipoproteins play a critical role in regulating the free drug fraction available for distribution, target engagement, metabolism, and elimination. Variations in protein binding resulting from physiological factors, disease states, age, drug interactions, and species differences can substantially alter drug efficacy and safety.
From a clinical perspective, a thorough understanding of drug-protein interactions is essential for optimizing dosage regimens, interpreting therapeutic drug monitoring results, and minimizing the risk of adverse drug reactions, particularly for highly protein-bound drugs and medications with narrow therapeutic indices. The growing emphasis on individualized medicine further highlights the importance of considering protein-binding variability when making therapeutic decisions in diverse patient populations.
Beyond its established pharmacological significance, knowledge of protein binding has important translational applications in drug development, precision pharmacotherapy, and pharmacokinetic modeling. Advances in analytical techniques, computational modeling, and artificial intelligence-based prediction tools are expected to improve the characterization and prediction of drug-protein interactions, thereby supporting more accurate dose optimization and personalized treatment strategies. Future research should focus on integrating protein-binding data with patient-specific factors to enhance therapeutic outcomes and promote safer, more effective medication use.
Acknowledgement
We would like to thank Dhaka University; Department of Pharmaceutical Chemistry authority for allowing us to use computer and library and other facilities. We also thank Department of Pharmacy, Independent University, Bangladesh and Department of Pharmacy, Atish Diponkar University of Science and Technology for their cooperation.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Permission to reproduce material from other sources
Not Applicable.
Clinical Trial Registration
This study does not involve any clinical trials.
Author contributions:
- Mohammad Shah Amran: Conceptualized the review topic and planned the main framework of the manuscript. Provided overall guidance, reviewed the manuscript multiple times, and made final corrections prior to submission.
- Fareha Anan Shristy: Accumulated relevant literature and selected appropriate articles from various sources. Assisted in organizing the reviewed articles and contributed to manuscript writing and revision. She has also drawn all the figures.
- Tashfiya Zaman Mouree: Read and summarized research papers and prepared the original draft based on the main framework of the manuscript.
- Saif Bin Salam Bondhon: Assisted in writing the original draft, compared findings from different studies, and contributed to structuring the review sections and revising the manuscript.
- Fahima Aktar: Verified reference articles, summarized key findings, and contributed to manuscript writing, revising and editing.
- Shaila Kabir: Confirmed critical information and contributed to the interpretation, structuring of findings, and overall manuscript review.
References
- Jusko WJ, Gretch M. Plasma and tissue protein binding and pharmacokinetics. Drug Metab Rev. 1976;5:43-140.
CrossRef - Blaschke TF. Protein binding and kinetics in liver disease. Clin Pharmacokinet. 1977;2:32-44.
CrossRef - Rowland M. Protein binding and drug clearance. Clin Pharmacokinet. 1984;9(Suppl 1):10-17.
CrossRef - Olson RE, Christ DD. Plasma protein binding of drugs. Annu Rep Med Chem. 1996;31:327-336.
CrossRef - Du Souich P, Verges J, Erill S. Plasma protein binding and pharmacological response. Clin Pharmacokinet. 1993;24(6):435-440.
CrossRef - Liu H, Chen P, Zhai X, et al. PPB-Affinity: Protein-protein binding affinity dataset for AI-based protein drug discovery. Sci Data. 2024;11(1):1316.
CrossRef - Cui M, Du Y. Native mass spectrometry for characterization of proteins binding with small molecules and application in drug discovery. TrAC Trends Anal Chem. 2024;174:117701.
CrossRef - Gupta S, Gupta A, Mukherjee M, Bose S, Sinha S. Chemical insights into oligonucleotide-protein binding for therapeutic applications. J Med Chem. 2025;68(10):9848-9863.
CrossRef - Courville J, Roupe K, Arold G. Re-discover the value of protein binding assessments in hepatic and renal impairment studies and its contributions in drug labels and dose decisions. Clin Transl Sci. 2024;17(5).
CrossRef - Al-Qassabi J, Tan SPF, Phonboon P, Galetin A, Rostami-Hodjegan A, Scotcher D. Facing the facts of altered plasma protein binding: Do current models correctly predict changes in fraction unbound in special populations? J Pharm Sci. 2024;113(6):1664-1673.
CrossRef - Heinen T, Fuhr U. Understanding plasma protein binding of drugs: Considering complexity and binding dynamics. J Pharm Sci. 2024;113(9):2994-2995.
CrossRef - Zhao H. Plasma protein binding as an optimizable parameter for in vivo efficacy. J Med Chem. 2025;68(11):12136-12140.
CrossRef - RadhaMahendran S, Dogra A, Mendhe D, Babu SBT, Dixit S, Singh SP. Machine learning for drug discovery: Predicting drug-protein binding affinities using graph convolutional networks. In: 2024 5th International Conference on Recent Trends in Computer Science and Technology (ICRTCST). IEEE; 2024:87-92.
CrossRef - Lindup WE, Orme MC. Clinical pharmacology: plasma protein binding of drugs. Br Med J. 1981;282(6259):212.
CrossRef - Trainor GL. The importance of plasma protein binding in drug discovery. Expert Opin Drug Discov. 2007;2(1):51-64.
CrossRef - Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. 2021;372. doi:10.1136/bmj.n71
CrossRef - Amran MS, Hossain MA. Study of protein binding of diltiazem in presence of omeprazole and ranitidine. J Bio-sci. 1998;6:115-121.
- Amran MS, Uzzaman MR, Hossain MA. Study of protein binding of diltiazem in presence of nifedipine and ketotifen fumarate. Rajshahi Univ Stud Part B. 1999;27:45-54.
- Amran MS, Hossain MA. Influence of ferrous sulphate on the protein binding of diltiazem in vitro. J Bangladesh Acad Sci. 1999;23(2):125-131.
- Amran MS, Bari AHMR, Hossain MA. The in vitro effects of ibuprofen and naproxen on the protein binding of diltiazem in aqueous medium. J Bio-sci. 2005;13:65-70.
- Mohiuddin M, Azam ATMZ, Amran MS, Hossain MA. In vitro effects of gliclazide and metformin on protein binding of caffeine in aqueous media. J Biol Sci. 2009;9(5):476-481.
CrossRef - Salam MA, Baki MA, Azam ATMZ, et al. In vitro and in vivo effects of glipizide and gliclazide on the protein binding, plasma concentration and biochemical parameters of ibuprofen. J Pharmacol Toxicol. 2009;4(8):307-313.
CrossRef - Siraji F, Azam ATMZ, Uddin MG, et al. In vitro effects of copper (II) and chromium (III) on protein binding of metronidazole and mebendazole. Int J Res Rev Pharm Appl Sci. 2012;2(5):916-929.
- Deepa KN, Hossain MK, Amran MS, Kabir S. In vitro model for studying interactions between ketorolac and omeprazole with bovine serum albumin by UV-spectroscopic method. Bangladesh Pharm J. 2014;17(1):92-98.
CrossRef - Koly SF, Sultan MZ, Rahman A, Kabir S, Amran MS. In vitro study of binding of aceclofenac and pantoprazole with bovine serum albumin by UV spectroscopic method. J Pharm Sci Emerg Drugs. 2015;3(2).
- Koly SF, Kundu SP, Kabir MS, Amran MS, Sultan MZ. Analysis of aceclofenac and bovine serum albumin interaction using fluorescence quenching method. EPMA J. 2015;6:24.
CrossRef - Karim SMR, Koly SF, Amran MS, Kabir S. In vivo studies of protein binding of ketorolac in rat model. Dhaka Univ J Pharm Sci. 2016;15(1):63-67.
CrossRef - Deepa KN, Sultan MZ, Amran MS, Kabir S. In vitro analysis of interaction between ketorolac tromethamine and bovine serum albumin using fluorescence spectroscopy. J Adv Med Pharm Sci. 2016;10(1):1-8.
CrossRef - Deepa KN, Kabir S, Amran MS. Fluorescence spectroscopic analysis of interaction between omeprazole and bovine serum albumin. World J Pharm Pharm Sci. 2016;5(9):16-25.
- Aktar F, Chowdhury RN, Akter F, Morshed N, Amran MS. Methodological advances in the study of drug-drug interactions. In: Pharmaceutical Science: New Insights and Developments. 2025. Manuscript No. 2025/BPR/6360.
CrossRef - Mohiuddin M, Amran MS, Hossain MA. In vivo effects of caffeine on hypoglycemic activity of gliclazide and metformin in healthy rats. Dhaka Univ J Pharm Sci. 2009;8(1):47-51.
CrossRef - Yotsuyanagi T, Ohta N, Futo T, Ito S, Chen DN, Ikeda K. Multiple and irreversible binding of cis-diamminedichloroplatinum(II) to human serum albumin and its effect on warfarin binding. Chem Pharm Bull (Tokyo). 1991;39:3003-3006.
CrossRef - Vuignier K, Schappler J, Veuthey JL, Carrupt PA, Martel S. Drug-protein binding: a critical review of analytical tools. Anal Bioanal Chem. 2010;398(1):53-66.
CrossRef - Singlas E. Protein Binding of Drugs: Definition, Modalities, Effects, Changes. F Hoffmann-La Roche & Co Ltd; 1987.
- Belpaire FM, Bogaert MG, Rosseneu M. Binding of beta-adrenoceptor blocking drugs to human serum albumin, alpha-1-acid glycoprotein, and human serum. Eur J Clin Pharmacol. 1982;22:253-256.
CrossRef - Routledge P. The plasma protein binding of basic drugs. Br J Clin Pharmacol. 1986;22(5):499-506.
CrossRef - Abedi F, Zarei B, Elyasi S. Albumin: A comprehensive review and practical guideline for clinical use. Eur J Clin Pharmacol. 2024;80(8):1151-1169.
CrossRef - Belinskaia DA, Jenkins RO, Goncharov NV. Albumin is an integrative protein of blood plasma and beyond. Int J Mol Sci. 2024;25(23):12627.
CrossRef - Yamasaki K, Chuang VTG, Maruyama T, Otagiri M. Albumin-drug interaction and its clinical implication. Biochim Biophys Acta. 2013;1830(12):5435-5443.
CrossRef - van der Sluijs P, Meijer DKF. Binding of drugs with a quaternary ammonium group to alpha-1-acid glycoprotein and asialo alpha-1-acid glycoprotein. J Pharmacol Exp Ther. 1985;234:703-707.
CrossRef - Barre J, Houin G, Rosenbaum J, Zini R, Dhumeaux D, Tillement JP. Decreased alpha-1-acid glycoprotein in liver cirrhosis: consequences for drug protein binding. Br J Clin Pharmacol. 1984;18:652-653.
CrossRef - Jonas A. Lipoprotein structure. In: Vance DE, Vance JE, eds. Biochemistry of Lipids, Lipoproteins and Membranes. Elsevier; 2002:483-504.
CrossRef - Wasan KM, Brocks DR, Lee SD, et al. Impact of lipoproteins on the biological activity and disposition of hydrophobic drugs: implications for drug discovery. Nat Rev Drug Discov. 2008;7(1):84-99.
CrossRef - Evans GH, Wilkinson GR, Shand DG. The disposition of propranolol IV: a dominant role for tissue uptake in the dose-dependent extraction of propranolol by the perfused rat liver. J Pharmacol Exp Ther. 1973;186:447.
CrossRef - Glazko AJ, Chang T. Diphenylhydantoin: absorption, distribution, and excretion. In: Woodbury DM, Penry JK, Schmidt RP, eds. Antiepileptic Drugs. Raven; 1972:127-136.
- Peterson RE, Wyngaarden JB. The miscible pool and turnover rate of hydrocortisone in man. J Clin Invest. 1956;35:552-561.
CrossRef - Levy G. Relationship between plasma protein binding, distribution, and anticoagulant action of dicumarol. Ann N Y Acad Sci. 1973;226:195-199.
CrossRef - Jusko WJ, Dittert LW. Integral coefficients of multicompartmental pharmacokinetic models. 1972;17:109-120.
CrossRef - Davis DR, Yeary RA. Bilirubin binding to hepatic Y and Z protein (ligandin): tissue bilirubin concentration in phenobarbital-treated Gunn rat. Proc Soc Exp Biol Med. 1975;148:9-13.
CrossRef - Lasser EC, Fan RS, Fujimagari T, Tripp WN. The significance of protein binding of contrast media in roentgen diagnosis. Am J Roentgenol Radium Ther Nucl Med. 1962;87:338-365.
- Piafsky KM. Disease-induced changes in the plasma binding of basic drugs. Clin Pharmacokinet.
CrossRef
- Wanat K. Biological barriers and the influence of protein binding on drug transport. Mol Biol Rep.
CrossRef
- Kober A, Jenner A, Sjöholm I. Differentiated effects of liver cirrhosis on the albumin binding sites for diazepam, salicylic acid, and warfarin. Biochem Pharmacol. 1978;27:2729-2735.
CrossRef - Huang Z, Ung T. Effect of alpha-1-acid glycoprotein binding on pharmacokinetics and pharmacodynamics. Curr Drug Metab.
CrossRef
- Abdallah YEH, Chahal S, Jamali F, Mahmoud SH. Drug-disease interaction: clinical consequences of inflammation on drugs action and disposition. J Pharm Pharm Sci. 2023;26:11137.
CrossRef - Oshikoya KA, Senbanjo IO. Pathophysiological changes that affect drug disposition in protein-energy malnourished children. Nutr Metab. 2009;6(1):50.
CrossRef - Ehrnebo M, Agurell S, Jalling B, Boreus LO. Age differences in drug binding by plasma proteins: studies on human foetuses, neonates, and adults. Eur J Clin Pharmacol. 1971;3:189-193.
CrossRef - Chignell CF, Vesell ES, Starkweather DK, Berlin CM. The binding of sulfaphenazole to fetal, neonatal, and adult human plasma albumin. Clin Pharmacol Ther. 1971;12:897-901.
CrossRef - Mather LE, Tucker GT, Pflug AE, Lindop MJ, Wilkinson G. Meperidine kinetics in man: intravenous injection in surgical patients and volunteers. Clin Pharmacol Ther. 1975;17:21-30.
CrossRef - Hadjian AJ, Chedin M, Cochet C, Chambaz EM. Cortisol binding to protein in plasma in the human neonate and infant. Pediatr Res. 1975;9:40-45.
CrossRef - Stout RJ. Some clinical evidence for a quantitative relationship between dose of relaxant and plasma protein. West Indian Med J. 1963;12:256.
- Payne RB, Little AJ, Williams RB, Milner JR. Interpretation of serum calcium in patients with abnormal serum proteins. Br Med J. 1973;4:643-646.
CrossRef - Thorp JM. Inter- and intraspecies differences in the binding of anionic compounds to albumin. Excerpta Med Int Congr Ser. 1972;254:98-109.
- Aoyagi T. Protein binding of rifampicin to different individual sera. Jpn J Antibiot. 1973;84:44-49.
- Schmidt DH, Butler VP. Immunological protection against digoxin toxicity. J Clin Invest. 1971;50:866-871.
CrossRef - Bluestone RI, Kippen I, Klinenberg JR. Effect of drugs on urate binding to plasma proteins. Br Med J. 1969;4:590.
CrossRef - Gogola D, Ratajczak SN, Gabor-Worwa E, et al. Implementation of a novel method for the determination of plasma protein binding of highly bound compounds using the equilibrium dialysis method coupled with extraction to the organic phase and its comparison to known methods. J Pharm Biomed Anal. 2025;255:116665.
CrossRef - Costa R, Ferreira C, Alves A, et al. Lipid-lowering statins and polyphenol-based supplementation: A scoping review on drug-food interaction potential. Front Pharmacol. 2025;16:1541871.
CrossRef - da Silva ECA, Costa MA, de Moura-Costa GF. Review of drug-food interactions based on medications used by users of a Basic Health Unit in Sarandi, Parana. Rev Uningá. 2025;62.
CrossRef - Gangadhar J, Meghana A, Naga Lakshmi A, Anusha G. A review on mechanisms and clinical effects of food-drug interactions. J Pharma Insights Res. 2025;3(5):244-251.
CrossRef - Perucca E. Pharmacological and therapeutic properties of valproate. CNS Drugs. 2002;16(10):695-714.
CrossRef - Luca C. Drug-drug interactions mediated by protein binding displacement. Published 2024.
- Gu CH, Kuldipkumar A, Chauhan H. Factors affecting drug absorption and disposition. In: Gu CH, Kuldipkumar A, Chauhan H, eds. Academic Press; 2024:499-516.
CrossRef - Bailey DG, Dresser GK, Arnold JMO. Grapefruit-medication interactions: forbidden fruit or avoidable consequences? 2013;185(4):309-316.
CrossRef - Dufour C, Dangles O. Flavonoid-serum albumin complexation: determination of binding constants and binding sites by fluorescence spectroscopy. Biochim Biophys Acta. 2005;1721:164-173.
CrossRef - Wong KY, Nie Z, Wong MS, Wang Y, Liu J. Metal-drug coordination nanoparticles and hydrogels for enhanced delivery. Adv Mater. 2024;36(26):2404053.
CrossRef - Petersen KU, Schmalix W, Pesic M, Stöhr T. Drug-drug interaction potential of remimazolam: CYP 450, transporters, and protein binding. Curr Drug Metab. 2024;25(4):266-275.
CrossRef - Azeem K, Irfan I, Rashid Q, Singh S, Patel R, Abid M. Recent updates on interaction studies and drug delivery of antimalarials with serum albumin proteins. Curr Med Chem. 2024;31(25):3925-3953.
CrossRef - Sulyok M, Hann S, Hartinger CG, Keppler BK, Stingeder G, Koellensperger G. Two-dimensional separation schemes for investigation of the interaction of an anticancer ruthenium(III) compound with plasma proteins. J Anal At Spectrom. 2005;20(9):856-863.
CrossRef - Niazi SK. Critical assessment of AI-based protein structure prediction: Fundamental challenges and future directions in drug discovery. Comput Struct Biotechnol Rep. 2025:100064.
CrossRef




