
Manuscript accepted on :21-11-2025
Published online on: 23-12-2025
Plagiarism Check: Yes
Reviewed by: Dr. Moumita Hazra
Second Review by: Dr. Hassan Shora
Final Approval by: Dr. Patorn Piromchai
Fabian Mok Ping Xiang 1 and Vasudeva Rao Avupati2,3*
and Vasudeva Rao Avupati2,3*
1School of Postgraduate Studies, IMU University (Formerly known as International Medical University), Kuala Lumpur, Malaysia.
2Department of Pharmaceutical Chemistry, School of Pharmacy, IMU University (Formerly known as International Medical University), Kuala Lumpur, Malaysia.
3Centre of Excellence for Bioactive Molecules & Drug Delivery (BMDD), Institute for Research, Development & Innovation (IRDI), IMU University (Formerly known as International Medical University), Kuala Lumpur, Malaysia.
Corresponding Author E-mail: vasudevaraoavupati@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/3322
Abstract
The increasing frequency of global travel and migration has intensified the spread of viral infections, including COVID-19 (SARS-CoV-2), Hepatitis C (Hepatitis C virus, HCV), acquired immunodeficiency syndrome (Human Immunodeficiency Virus, HIV), Dengue fever (Dengue virus, DENV), and Zika fever (Zika virus, ZIKV). These global health challenges underscore the urgent need for effective antiviral therapeutics. Viral protease enzymes play a vital role in the viral replication cycle, making them promising molecular targets for therapeutic intervention. In this study, structure-based pharmacophore modeling (SBPM) and molecular docking approaches were employed to identify potential protease inhibitors against multiple viral proteases. Pharmacophore models were generated for five viral targets obtained from the RCSB Protein Data Bank, followed by virtual screening of compound libraries from the ZINC and Molport databases. Top-ranked virtual hits were subsequently docked using iGEMDOCK to evaluate binding affinities and molecular interaction profiles, while physicochemical and pharmacokinetic properties were assessed using the SWISSADME web tool. The findings revealed several promising virtual hits exhibiting strong and stable binding interactions at the protease active sites, alongside favorable drug-likeness profiles. These results provide a valuable foundation for future in vitro validation, hit-to-lead optimization, and the potential development of novel antiviral agents targeting multiple viral proteases.
Keywords
Dengue virus (DENV); Drug discovery; Hepatitis C virus (HCV); Human Immunodeficiency Virus (HIV); Molecular docking; SARS-CoV-2; Structure-based pharmacophore modeling (SBPM); Viral protease inhibitors; Virtual screening; Zika virus (ZIKV)
Download this article as:| Copy the following to cite this article: Xiang F. M. P, Avupati V. R. Identification of Inhibitors of Viral Proteases by Virtual Screening: A Structure-Based Pharmacophore Modelling (SBPM) and Molecular Docking Studies. Biomed Pharmacol J 2025;18(4). |
| Copy the following to cite this URL: Xiang F. M. P, Avupati V. R. Identification of Inhibitors of Viral Proteases by Virtual Screening: A Structure-Based Pharmacophore Modelling (SBPM) and Molecular Docking Studies. Biomed Pharmacol J 2025;18(4). Available from: https://bit.ly/4qnWlvB |
Introduction
The field of drug discovery is characterized by a continuous and dynamic process involving multiple stages, from target identification to preclinical development, before eventual clinical evaluation. This process traditionally involves laborious experimental screening of compound libraries to identify potential drug candidates. However, with the completion of the human genome project, there has been a surge in the number of potential therapeutic targets for drug discovery, necessitating more efficient and innovative approaches to accelerate the discovery of novel antiviral drugs. Traditional methods are time-consuming and resource-intensive, hence the adoption of computational techniques.1 for complimenting and expediting the drug discovery process.2 The study leverages advancements in structural biology, computational modeling, and bioinformatics to facilitate the identification of potential inhibitors. Integration of artificial intelligence and machine learning enhances screening protocols. Challenges such as drug resistance and off-target effects persist, necessitating continued research efforts.3 The emergence of viral infections, such as COVID-19 (SARS-CoV-2), Hepatitis C (Hepatitis C virus, HCV), Acquired Immunodeficiency Syndrome (Human Immunodeficiency Virus, HIV), Dengue fever (Dengue virus, DENV), and Zika fever (Zika virus, ZIKV). presents a significant global health challenge. Viral proteases play crucial roles in the replication and maturation of these viruses, making them attractive targets for drug discovery. However, the identification of potent and selective inhibitors against viral proteases remains a daunting task. Traditional drug discovery methods often require extensive experimental screening of compound libraries, which is time-consuming and resource intensive. Hence, there is a pressing need for innovative computational approaches to expedite the discovery of novel antiviral drugs targeting viral proteases. Advancements in structural biology, computational modeling, and bioinformatics have enabled a deeper understanding of viral proteases’ structure-function relationships and mechanisms of action, facilitating the identification of potential inhibitors with enhanced efficacy and specificity. Traditional methods of drug discovery are time-consuming and resource-intensive, so the adoption of innovative computational techniques is crucial. Structure-based drug design combined with high-throughput screening methodologies offers a streamlined approach for identifying lead compounds with promising antiviral activity. The integration of artificial intelligence and machine learning algorithms has revolutionized drug discovery by enhancing the predictive power and efficiency of screening protocols. Challenges such as drug resistance and off-target effects persist, necessitating ongoing research and development efforts. Drug repurposing, particularly leveraging existing compounds as protease inhibitors, is a viable strategy for circumventing these obstacles while minimizing development costs and timelines. Thus, pursuing innovative computational approaches in antiviral drug discovery is essential to combat viral infections effectively. 4
Coronaviruses (CoVs) have garnered global attention due to their potential for causing severe respiratory illnesses, exemplified by outbreaks such as Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and the ongoing pandemic caused by SARS-CoV-2.5,6 The role of SARS-CoV-2 proteases in viral replication and host immune modulation is pivotal in the ongoing COVID-19 pandemic.7,8 These proteases, including the main protease (Mpro) and the papain-like protease (PLpro), facilitate viral polyprotein processing and manipulate host immune responses, contributing to viral immune evasion and disease severity. Consequently, targeting these proteases has emerged as a promising strategy for antiviral drug development. Inhibitors disrupting their catalytic activity have shown efficacy in suppressing viral replication and improving clinical outcomes. Structural elucidation of SARS-CoV-2 proteases through techniques like x-ray crystallography and cryo-electron microscopy has guided the rational design of inhibitors, enhancing their potency and specificity. However, challenges such as developing broad-spectrum inhibitors against diverse viral strains, minimizing off-target effects, and ensuring drug affordability and accessibility persist, underscoring the importance of continued research efforts in combatting COVID-19.
Hepatitis C virus (HCV) stands as a significant contributor to chronic liver disease, culminating in conditions like cirrhosis and hepatocellular carcinoma. The NS3/4A protease, pivotal in HCV replication, orchestrates viral polyprotein cleavage, facilitating the synthesis of crucial functional proteins crucial for viral assembly and infectivity. 9 Moreover, by targeting key host proteins linked to innate immunity, this protease also manipulates host immune responses, aiding viral evasion of defenses and contributing to disease progression.10,11 Antiviral drug discovery has homed in on this protease, yielding inhibitors that show promise in halting viral replication and ameliorating clinical outcomes in HCV-infected individuals. These inhibitors disrupt the catalytic activity of NS3/4A, impeding viral polyprotein processing and replication, with structural studies guiding the development of more potent and specific inhibitors. Techniques such as X-ray crystallography and cryo-electron microscopy have unveiled the protease’s three-dimensional structure and its complexes with inhibitors at atomic resolution, laying the groundwork for structure-based drug design and optimization of lead compounds against HCV NS3/4A. Challenges persist in developing inhibitors effective across diverse HCV genotypes and minimizing off-target effects and toxicity. Additionally, ensuring affordable access to these drugs, particularly in regions heavily burdened by HCV-related liver disease, is critical for global health improvement.12
Human Immunodeficiency Virus type 1 (HIV-1) poses a formidable global health challenge, leading to acquired immunodeficiency syndrome (AIDS).13 Its protease is instrumental in viral replication, cleaving the viral polyprotein and influencing host immune responses, thus aiding viral evasion, and contributing to disease progression.14,15 Antiviral drug discovery has focused on inhibiting HIV-1 protease, yielding protease inhibitors that revolutionized HIV treatment by reducing viral load and enhancing clinical outcomes.16–18 Structural studies have provided insights into the protease’s mechanism, guiding the development of potent inhibitors through techniques like X-ray crystallography and cryo-electron microscopy.15 Challenges persist, including the need for inhibitors with improved pharmacokinetics and reduced resistance, alongside efforts to enhance drug safety and accessibility, especially in resource-limited regions, to bolster global HIV control efforts. 18
The Dengue virus (DENV) poses a formidable global health threat, with millions of infections yearly. Central to DENV replication is the NS2B-NS3 protease complex, pivotal for polyprotein processing and replication, and influencing host immune responses, thus contributing to disease severity.19,20 Inhibitors targeting NS2B-NS3 have shown promise in suppressing viral replication and improving clinical outcomes, impeding viral polyprotein processing and replication. Structural elucidation of NS2B-NS3 has guided the development of potent inhibitors. 21–24 Challenges persist, including the need for broad-spectrum inhibitors, minimizing off-target effects, and ensuring access to affordable antiviral drugs in DENV-endemic regions. 25–27
Zika virus (ZIKV) has emerged as a significant public health concern due to its association with neurological complications such as microcephaly and Guillain-Barré syndrome.28 The NS2B-NS3 protease complex of ZIKV plays a crucial role in viral replication by cleaving the viral polyprotein, facilitating the production of individual functional proteins necessary for viral assembly and infectivity, while also modulating host immune responses by cleaving key host proteins involved in innate immunity, enabling viral evasion of host defenses and contributing to disease pathogenesis.29,30 The essential role of the NS2B-NS3 protease in ZIKV replication makes it an attractive target for antiviral drug development, with inhibitors showing promise in suppressing viral replication and improving clinical outcomes in infected individuals. Structural elucidation of the Zika virus NS2B-NS3 protease has provided valuable insights into its catalytic mechanism and substrate specificity, facilitating the design of novel therapeutics through techniques such as X-ray crystallography and cryo-electron microscopy (cryo-EM).31,32 Despite progress, challenges remain in developing inhibitors with broad-spectrum activity against diverse ZIKV strains and related flaviviruses, while efforts to minimize off-target effects, toxicity, and increase access to affordable antiviral drugs are vital for mitigating the global impact of ZIKV infection and associated neurological complications.33–35
This study aims to develop structure-based pharmacophore models (SBPMs) derived from the binding site regions of selected disease-specific viral proteases to facilitate the identification of potential inhibitors. The generated SBPMs will be employed for virtual screening of compound libraries to identify stable virtual hits for each viral protease. Subsequently, molecular docking studies will be performed to predict and evaluate the binding profiles of these hits at the target binding sites. The research further seeks to elucidate the key structural features responsible for selective inhibitor binding to viral proteases. It is hypothesized that the virtual hits identified through SBPMs and molecular docking analyses will exhibit in silico stable binding profiles at the putative target sites comparable to, or better than, those of known specific inhibitors. The novelty of this study lies in its two-tiered screening strategy, wherein the initial hits prioritized based on SBPMs for specific viral proteases are subsequently re-ranked through molecular docking studies making this approach distinct from conventional direct docking-based virtual screening protocols.
Materials and Methods
Target Selection
A systematic, multi-step selection process was applied to identify suitable viral protease targets. Five proteases were chosen for this study: Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV-2) protease, Hepatitis C virus (HCV) NS3/4A protease, HIV-1 protease, Dengue virus NS2B–NS3 protease, and Zika virus NS2B–NS3 protease. The three-dimensional (3D) structures of these targets were retrieved from the RCSB Protein Data Bank (www.rcsb.org) and PDBsum (https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/) databases based on the following criteria: (i) availability of X-ray crystallographic data with acceptable resolution, (ii) presence of a co-crystallized ligand complex to allow interaction analysis, (iii) absence of chain breaks in the protein sequence, and (iv) high-quality Ramachandran plot statistics, with >90% residues in the most favorable region, <10% in additionally and generously allowed regions, and <0.1% in disallowed regions. All heteroatoms were removed from the selected protein structures using UCSF Chimera (https://www.cgl.ucsf.edu/chimera/).
Structure-Based Pharmacophore Modelling (SBPM) and Virtual Screening
The selected X-ray derived 3D structures of the viral proteases were subjected to structure-based pharmacophore modeling using the Pharmit web server (https://pharmit.csb.pitt.edu/). Pharmit analyzes ligand–protein complexes to identify pharmacophoric features such as hydrophobic centers, hydrogen bond donors and acceptors, and aromatic rings. For virtual screening, the following filtering parameters were applied: a maximum of 1000 hits for hit reduction and the “beyond rule of five” (bRo5) filter for hit screening. This criterion was used instead of Lipinski’s Rule of Five, as protease inhibitors often possess larger and more complex structures than typical small-molecule drugs. Beyond Rule of Five (bRo5) filter parameters: Molecular weight < 500, cLogP between 0 and 7.5, Hydrogen bond donors ≤ 5, Hydrogen bond acceptors ≤ 10, Polar surface area ≤ 200 Ų, Rotatable bonds ≤ 20. Hits were ranked based on root mean square deviation (RMSD) and minimized RMSD (mRMSD) values, representing 3D conformational fit to the pharmacophore. The maximum score threshold was set to 0, and the optimal mRMSD was ≤ 2 Å. Pharmit-generated pharmacophore models were used to screen integrated chemical databases, including ZINC, ChEMBL34, PubChem, and MolPort respectively. The best-hit molecules were exported in SDF format for further preparation and docking analysis.
Molecular Docking
Ligand structures were drawn and energy-minimized using ChemDraw and Chem3D 21.0.0 software and saved in SDF format. Binding site analysis was performed based on co-crystallised and bioactive ligands and solvent molecules were removed from the PDB structures using UCSF Chimera. Molecular docking was performed using the open-source software iGEMDOCK (http://gemdock.life.nctu.edu.tw/igemdock.php). This tool generates protein–compound complexes, profiles target binding interactions, and ranks ligands based on total binding energy, van der Waals energy, and hydrogen bonding contributions. The resulting binding poses, and molecular interactions were analyzed using PoseView by ProteinPlus (https://proteins.plus), which provided two-dimensional representations of hydrogen bonds, hydrophobic interactions, and π–π stacking between the hit compounds and the active site residues of each protease.
ADME Prediction
The pharmacokinetic and physicochemical properties of the virtual hits were predicted using the SWISSADME web tool (http://www.swissadme.ch/). The SMILES notations of the compounds were entered to calculate key descriptors, including topological polar surface area (TPSA), molecular weight, number of heavy atoms, hydrogen bond donors and acceptors, and rotatable bonds. Predicted lipophilicity (iLOGP) and solubility (Log S, ESOL model) were used to assess water solubility and membrane permeability. Gastrointestinal (GI) absorption and blood–brain barrier (BBB) permeability were also evaluated. In addition, Lipinski’s score, lead-likeness, and synthetic accessibility were determined to facilitate comparison among the hits and the reference inhibitor, and to prioritize compounds with favorable drug-like and synthetic characteristics for further lead optimization studies.
Results
Target selection
The selection of appropriate viral protease targets is a crucial step in structure-based drug design, as it determines the accuracy and biological relevance of subsequent pharmacophore modeling and virtual screening studies. As part of this study, a set of five disease-specific viral proteases were chosen based on their established roles in viral replication and pathogenesis, as well as the availability of high-resolution crystal structures in the RCSB Protein Data Bank (PDB). Each selected protease represents a key therapeutic target for a major viral infection of global concern. The details of the selected protease targets, including their corresponding PDB IDs, are presented in Table 1 below.
Table 1: Structural details of selected viral protease targets used in this study.
| PDB ID | Viral protease | Ligand |
| 8HUR | SARS-CoV-2 main protease (Mpro)36 | Ensitrelvir |
| 4WF8 | HCV protease37 | Asunaprevir |
| 3SPK | HIV-1 protease38 | Tipranavir |
| 2FOM | DENV protease39 | *Anthracene-1,2,7,9-tetraol22 |
| 5H4I | ZIKV protease40 | *N-(5-nitro-6-(piperazin-1-yl) pyridin-2-yl)-1H-indazol-5-amine41 |
* As no co-crystallized ligand was available, the binding analysis was performed using a bioactive ligand reported in the literature.
The key chemical features generated from the structure-based pharmacophore models (SBPMs) of the selected viral proteases are summarized in Table 2 below and the models shown in Figure 1. These features represent the essential interactions within the active sites that govern ligand recognition and binding specificity.
Table 2: Chemical Features of Viral Protease-Based Structure-Based Pharmacophore Models (SBPMs)
| PDB ID | Structure-Based Pharmacophores |
| 8HUR | 4 Hydrogen acceptors, 1 Hydrogen donor 1 Aromatic ring |
| 4WF8 | 1 Aromatic, 4 Hydrogen acceptors, 1 Hydrogen donor |
| 3SPK | 3 Hydrogen acceptors, 2 Hydrogen donors, 1 Hydrophobic |
| 2FOM | 4 Hydrogen acceptors, 4 Hydrogen donors |
| 5H4I | 1 Positive ion, 1 Negative ion, 4 Hydrogen acceptors, 2 Hydrogen donors |
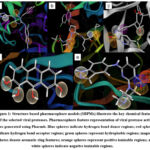 |
Figure 1: Structure-based pharmacophore models (SBPMs) illustrate the key chemical features of the selected viral proteases. Pharmacophore feature representation of viral protease active sites generated using Pharmit. |
Table 3: Docking scores and key target–ligand binding interactions of the top three virtual hits and the reference inhibitor against SARS-CoV-2 main protease (PDB ID: 8HUR).
| Hit molecule | Docking Score (kcal/mol) | Target-ligand binding interactions |
| Molport-046-596-817 | -142.972 | His41: hydrogen bond, hydrophobic interactionMet165: hydrophobic interaction
Asn142: hydrophobic interaction |
| Molport-002-530-884 | -140.026 | Glu166: hydrogen bond, ionic interaction, hydrophobic interactionHis41: pi-pi interaction, hydrophobic interaction
Met165: hydrophobic interaction |
| Molport-002-530-949 | -138.131 | Ser144: hydrogen bondGlu166: hydrogen bond
His41: pi-pi interaction Met165: hydrophobic interaction Glu189: hydrophobic interaction His41: hydrophobic interaction |
| Ensitrelvir(Co-crystallised ligand) | -140.431 | His163: hydrogen bondGly143: hydrogen bond
His41: pi-pi interaction Thr26: hydrophobic interaction Cys145: hydrophobic interaction |
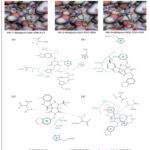 |
Figure 2: Comparison of the top three virtual hits showing (A) 2D molecular structures, (B) 3D docking poses, and (C) 2D ligand–protein interaction diagrams for the reference inhibitor, Hit-1, Hit-2, and Hit-3, respectively. |
Table 4: Docking scores and key target–ligand binding interactions of the top three virtual hits and the reference inhibitor against Hepatitis C viral protease (PDB ID: 4WF8).
| Hit molecule | Docking Score (kcal/mol) | Target-ligand binding interactions |
| Molport-020-311-760 | -139.978 | His1057: hydrophobic interactionAla1156: hydrophobic interaction
His1057: pi-pi interaction |
| Molport-020-309-674 | -135.824 | His1057: pi-pi interaction, hydrophobic interactionAsp1079: hydrophobic interaction
Val1078: hydrogen bond |
| Molport-047-017-485 | -127.644 | Ser1139: hydrogen bondLys1136: hydrophobic interaction
Ala1156: hydrophobic interaction His1057: hydrophobic interaction |
| Asunaprevir(Co-crystallised ligand) | -123.328 | Asp1081: hydrophobic interactionArg1155: hydrophobic interaction, hydrogen bond
Ala1156: hydrophobic interaction Ala1157: hydrophobic interaction, hydrogen bond His1057: hydrophobic interaction, hydrogen bond Gly1137: hydrogen bond Ser1139: hydrogen bond |
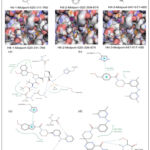 |
Figure 3: Comparison of the top three virtual hits showing (A) 2D molecular structures, (B) 3D docking poses, and (C) 2D ligand–protein interaction diagrams for the reference inhibitor, Hit-1, Hit-2, and Hit-3, respectively. |
Table 5: Docking scores and key target–ligand binding interactions of the top three virtual hits and the reference inhibitor against HIV-1 viral protease (PDB ID: 3SPK)
| Hit molecule | Docking Score (kcal/mol) | Target-ligand binding interactions |
| Molport-008-344-770 | -135.824 | Asn25A: hydrogen bondAsn25B: hydrogen bond
Thr82: hydrophobic interaction Leu23: hydrophobic interaction Pro81: hydrophobic interaction Ala28: hydrophobic interaction |
| Molport-023-246-998 | -127.644 | Asp30: hydrogen bondIle50: hydrogen bond |
| Molport-001-545-102 | -127.644 | Ile50: hydrogen bondAsn25: hydrogen bond
Gly48: hydrogen bond Asp29: hydrogen bond Arg8: pi-cation interaction, hydrophobic interaction Leu23: hydrophobic interaction Gly49: hydrophobic interaction |
| Tipranavir(Co-crystallised ligand) | -138.524 | Ile50A: hydrogen bondIle50B: hydrogen bond
Thr82: hydrophobic interaction Pro81: hydrophobic interaction Ala28: hydrophobic interaction Ile50: hydrophobic interaction Asn25A: hydrogen bond Asn25B: hydrogen bond Asp30: hydrogen bond Asp29: hydrogen bond Thr82: hydrophobic interaction Leu23: hydrophobic interaction |
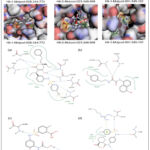 |
Figure 4: Comparison of the top three virtual hits showing (A) 2D molecular structures, (B) 3D docking poses, and (C) 2D ligand–protein interaction diagrams for the reference inhibitor, Hit-1, Hit-2, and Hit-3, respectively. |
Table 6: Docking scores and key target–ligand binding interactions of the top three virtual hits and the reference inhibitor against DENV protease (PDB ID: 2FOW).
| Hit molecule | Docking Score (kcal/mol) | Target-ligand binding interactions |
| Molport-044-727-279 | -121.0420 | Gly153: hydrogen bondGly151: hydrogen bond
Tyr161: pi-pi interaction His51: hydrophobic interaction Pro132: hydrophobic interaction Leu128: hydrophobic interaction |
| Molport-000-808-297 | -118.6330 | Tyr161: pi-pi interaction, hydrophobic interactionTyr150: hydrophobic interaction
His51: hydrophobic interaction Leu128: hydrophobic interaction Pro132: hydrophobic interaction Gly151: hydrogen bond Gly153: hydrogen bond |
| Molport-001-740-947 | -116.9030 | Tyr161: pi-pi interactionGly153: hydrogen bond
Ser135: hydrogen bond His51: hydrophobic interaction |
| Anthracene-1,2,7,9-tetraol(Bioactive ligand) | -90.1099 | Ser135: hydrogen bondGly153: hydrophobic interaction
Gly151: hydrogen bond |
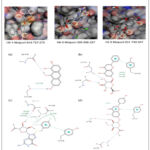 |
Figure 5: Comparison of the top three virtual hits showing (A) 2D molecular structures, (B) 3D docking poses, and (C) 2D ligand–protein interaction diagrams for the reference inhibitor, Hit-1, Hit-2, and Hit-3, respectively. |
Table 7: Docking scores and key target–ligand binding interactions of the top three virtual hits and the reference inhibitor against ZIKV protease (PDB ID: 5H4I).
| Hit molecule | Docking Score (kcal/mol) | Target-ligand binding interactions |
| ZINC000015953149 | -122.6440 | His51: hydrophobic interaction and pi-pi interactionAla132: hydrophobic interaction
Tyr161: hydrophobic interaction Asp83: hydrogen bond |
| ZINC00004825072 | -118.4590 | Tyr161: hydrogen bond, hydrophobic interactionHis51: hydrophobic interaction |
| Molport-002-888-347 | -117.7430 | Ala132: hydrophobic interactionHis51: hydrophobic interaction
Tyr161: hydrophobic interaction |
| N-(5-nitro-6-(piperazin-1-yl) pyridin-2-yl)-1H-indazol-5-amine(Bioactive ligand) | -99.415 | Tyr161: pi-pi interaction and hydrophobic interactionTyr150: hydrogen bond
Ala132: hydrophobic interaction |
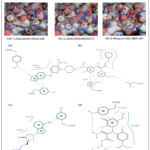 |
Figure 6: Comparison of the top three virtual hits showing (A) 2D molecular structures, (B) 3D docking poses, and (C) 2D ligand–protein interaction diagrams for the reference inhibitor, Hit-1, Hit-2, and Hit-3, respectively. |
Discussion
The application of a structure-based pharmacophore model (SBPM), derived from the SARS-CoV-2 main protease (Mpro) and its crystallized ligand, for virtual screening led to the identification of the virtual hits: Molport-002-527-96, Molport-002-528-694, Molport-002-530-593, Molport-002-530-797, Molport-002-530-884, Molport-002-530-890, Molport-002-530-949, Molport-002-531-829, and Molport-046-596-817 respectively. Subsequent, molecular docking studies against SARs-CoV-2 main protease (PDB ID: 8HUR) revealed strong hydrophilic and hydrophobic interactions between the ligand and the amino acid residues of the protease binding site. Ensitrelvir, used as the reference inhibitor, formed stable interactions with the active site residues His163, Gly143, Ser144, Cys145, and Thr26, confirming its established binding pattern within the protease pocket. Two-dimensional (2D) visualization of the ten screened hits demonstrated multiple hydrophilic interactions with amino acid residues including Leu205, Pro293, Phe3, Phe291, Trp207, Gly299, Ala206, Val212, Asp295, Val297, Ala210, Thr292, Phe4, Leu253, Cys300, and Arg298. The hydrophilic interactions primarily involved R–NH donor groups in the ligands forming hydrogen bonds with C=O acceptor groups of the protein backbone. π–π stacking interactions were also observed between the aromatic rings of the hit compounds and the aromatic side chains of amino acid residues, particularly those containing imidazole (His41), phenyl (Phe), and indole (Trp) groups. The triazole functional groups within the hit structures contributed strongly to these hydrophilic interactions, due to the presence of nitrogen atoms that act as hydrogen bond acceptors and the aromatic character that facilitates π–π stacking. The hydrophobic interactions were mainly attributed to substituents on the aromatic rings, which enhance contact with the enzyme’s hydrophobic pocket. Overall, the triazinone and triazole moieties present in both Ensitrelvir and the top hits accounted for the majority of the hydrophilic interactions through their carbonyl and nitrogen atoms. Among the docked hits, the top three compounds with the best binding energies were: Molport-046-596-817 (−142.972 kcal/mol), Molport-002-530-884 (−140.026 kcal/mol), Molport-002-530-949 (−138.131 kcal/mol), All three virtual hits demonstrated strong binding affinity and fit optimally within the protease active-site pocket, showing theoretical binding poses highly comparable to that of Ensitrelvir. Each of the three hits contained aromatic rings that facilitated π–π interactions with amino acid residues such as His41, and shared interactions with Met165 and His41, residues known to be critical for catalytic activity in the protease. Structurally, all three compounds featured tricyclic or fused polycyclic ring systems, including lactam rings (cyclic amides), which are characteristic of protease inhibitors. These rigid frameworks enhance binding affinity by improving shape complementarity with the enzyme pocket. Multiple amide bonds within the compounds serve as both hydrogen bond donors and acceptors, mimicking the peptide backbone of natural substrates and indicating potential activity like serine or cysteine protease inhibitors. Additionally, the presence of methylenedioxyphenyl groups (–O–CH₂–O–) attached to aromatic rings enhanced binding to hydrophobic regions within the enzyme’s active site and contributed to π–π stacking interactions. The overall planarity of these structures facilitated optimal fitting within the protease binding pocket (Table 3 and Figure 2). The incorporation of alkyl substituents and phenyl rings further contributed to membrane permeability and improved predicted bioavailability of the virtual hits. The ADME properties of the three selected hits were predicted using SWISSADME. None of these compounds violated the relaxed “beyond rule of five” criteria applied during the Pharmit filtering stage, suggesting that they all possess acceptable drug-like characteristics. For comparison, the reference inhibitor, Ensitrelvir, showed a lipophilicity (iLogP) value of 3.22, which is within the same range as the selected hits. Structurally, Ensitrelvir is a heterocyclic molecule with several important chemical features. It contains a methylated purine group that supports base-pair recognition, while its bicyclic urea core composed of two carbonyl and three nitrogen atoms provides molecular rigidity and planarity. The carbonyl groups enable hydrogen bonding, which plays a key role in ligand–protein interactions. Additionally, the substituted aryl amide ring with chlorine and a methylated nitrogen atom enhances lipophilicity and promotes π–π stacking interactions. The trifluoromethylphenyl side chain further contributes to stability and binding through van der Waals and dipolar interactions. Among the three hits, Molport-046-596-817 was predicted to have the lowest solubility (Log S = −4.44), while Molport-002-530-884 and Molport-002-530-949 displayed moderate solubility. Ensitrelvir, in comparison, had the lowest solubility overall (Log S = −4.84). All three hits achieved good Lipinski scores, indicating that they are likely to have favourable oral bioavailability. The predicted gastrointestinal (GI) absorption was high for both Ensitrelvir and most hits, except for Molport-002-530-949, which showed lower predicted GI absorption. Regarding synthetic feasibility, Molport-002-530-884 had the highest synthetic accessibility score (4.96), suggesting that it may be more complex to synthesise compared to Molport-046-596-817 and Molport-002-530-949. The reference inhibitor, Ensitrelvir, had a moderate synthetic accessibility score of 3.56. These results align well with recent developments in the targeting of SARS-CoV-2 main protease (Mᵖʳᵒ). A group of researchers used a combined ligand- and structure-based virtual screening protocol and screened ~200 million compounds, and emphasised the importance of hydrogen-bond donors/acceptors plus aromatic/π-interaction features in the pharmacophore design.42 In another synthetic research study unfolded the importance of triazole-piperazine derivatives that achieved sub-micromolar Mᵖʳᵒ inhibition (IC₅₀ ≈ 1.9 µM) and favourable liver microsome stability.43 In this context, the triazole/triazinone features, the observed H-bonding to Gly143, Ser144, Cys145, and π-π stacking with His41 are consistent with the reported literature.44
The application of a structure-based pharmacophore model (SBPM), derived from the Hepatitis C viral protease and its crystallized ligand, for virtual screening led to the identification of the virtual hits: Molport-003-895-473, Molport-005-131-122, Molport-010-740-325, Molport-010-819-952, Molport-010-819-959, Molport-020-309-674, Molport-020-311-760, Molport-028-743-611, Molport-047-008-406, and Molport-047-017-485. Subsequent, molecular docking studies against HCV NS3 protease (PDB ID: 4WF8) revealed strong hydrophilic and hydrophobic interactions between the selected ligands and the key amino acid residues within the catalytic binding pocket. Ensitrelvir, used as a reference inhibitor, formed π–π stacking and hydrophobic interactions with His1057 and Ala1156, validating the reliability of the docking approach and serving as a structural benchmark for comparison. Among the screened hits, three compounds Molport-020-311-760, Molport-020-309-674, and Molport-047-017-485 demonstrated highly favorable binding orientations and interaction energies within the protease pocket. Two-dimensional (2D) visualization of the ligand–protein interactions revealed multiple hydrophilic and hydrophobic contacts involving residues His1057, Ala1156, Asp1079, Val1078, Ser1139, Lys1136, and Ala1156. The hydrophilic interactions were primarily mediated through hydrogen bonding between amide, carbonyl, and heteroatom groups in the ligands and backbone carbonyl or side-chain donor groups of the protein. π–π stacking interactions were prominently observed between aromatic rings in the hit compounds and the imidazole side chain of His1057, supporting stable non-covalent anchoring within the protease active site. Additionally, polar substituents on aromatic rings facilitated directional hydrogen bonding with residues such as Val1078 and Ser1139, enhancing the overall binding complementarity of the ligands. The hydrophobic interactions were mainly contributed by alkyl and phenyl substituents, which enhanced contact with the enzyme’s hydrophobic sub-pocket near Ala1156 and Asp1079. Among the three hits, Molport-020-311-760 exhibited the strongest binding affinity, with the lowest docking energy (−139.978 kcal/mol) and minimal structural deviation (mRMSD = 1.741 Å). This compound showed predominant π–π stacking with His1057 and favorable hydrophobic packing with Ala1156, suggesting a stable and energetically optimized binding pose. The planar aromatic core of Molport-020-311-760 contributed to efficient shape complementarity and π–π overlap, while its limited polar surface (TPSA = 89.47 Ų) favored deep binding pocket accommodation. Molport-020-309-674, with a docking energy of −135.824 kcal/mol, showed a balanced interaction profile involving π–π stacking with His1057, hydrogen bonding with Val1078, and hydrophobic contacts with Asp1079 and Ala1156. The methoxy-substituted aromatic ring in this compound likely enhanced hydrogen-bonding potential and fine-tuned lipophilicity, contributing to improved solubility and stable docking orientation. Molport-047-017-485, while exhibiting a slightly weaker binding energy (−127.644 kcal/mol), displayed strong hydrophilic interactions through hydrogen bonding with Ser1139 and hydrophobic contacts with Lys1136, His1057, and Ala1156. The presence of multiple heteroatoms within its aromatic and amine functionalities enabled extensive hydrogen bonding, while its higher topological polar surface area (TPSA = 113 Ų) reflected increased polarity and solvation potential. Structurally, all three hits contained rigid aromatic scaffolds capable of sustaining π–π stacking and hydrophobic complementarity, features typical of protease inhibitors. The presence of amide and heterocyclic linkages in each compound provided multiple hydrogen-bond donors and acceptors, allowing interactions that mimic peptide backbone recognition within the catalytic site. The planar aromatic systems enhanced fitting within the active-site cleft, while the alkyl and phenyl substituents contributed to hydrophobic stabilization and potential membrane permeability. The observed interaction patterns and docking geometries suggest that all three ligands occupy the protease binding groove in a manner similar to known NS3 inhibitors, engaging key residues associated with catalytic function (Table 4 and Figure 3). The physicochemical and ADME profiles of these compounds, predicted using SwissADME, indicated favorable drug-like characteristics. None of the three hits violated Lipinski’s rule of five, and all showed iLogP values between 3.19 and 3.84 comparable to Ensitrelvir (iLogP = 3.22) indicating balanced lipophilicity for potential oral bioavailability. The calculated solubility (Log S) values ranged from −4.14 to −4.87, suggesting moderate solubility suitable for early-stage in vitro evaluation. Molport-047-017-485 exhibited the lowest solubility (−4.87) but the best synthetic accessibility (SA = 3.59), whereas Molport-020-311-760 demonstrated the most potent binding energy with acceptable solubility (−4.14) and a manageable synthetic complexity (SA = 3.98). All compounds were predicted to have high gastrointestinal (GI) absorption and low probability of blood–brain barrier penetration, consistent with the desired pharmacokinetic profile of peripheral protease inhibitors. Overall, Molport-020-311-760 emerged as the most promising hit based on its strong binding energy, favorable lipophilicity, and excellent geometric complementarity with the protease binding site. Molport-020-309-674 presented a balanced profile of solubility, stability, and binding interactions, making it an attractive candidate for further optimization. Molport-047-017-485, with its high pharmacophore fit and synthetic accessibility, represents a valuable scaffold for structural modification to enhance π–π stacking interactions and potency. The consistent presence of aromatic and amide functionalities across all three hits highlights their potential as lead-like scaffolds for further development of HCV NS3 protease inhibitors. These observations align with current strategies in targeting the NS3/4A protease of Hepatitis C virus, where aromatic scaffolds capable of π–π stacking and amide-rich linkages continue to strengthen high-affinity binding. A recent study modelling genotype-3 variants of NS3/4A used pharmacophore-assisted covalent docking and molecular dynamics to show that His57 (canonical numbering) engages in persistent π–π interactions, and that variations in hydrophobic groove residues affect ligand binding.45 Additional computational work demonstrates that virtual screening against NS3/4A reveals conserved interaction motifs: hydrogen bonds to residues in the catalytic loop (e.g., Asp81/Ser139) and occupation of adjacent hydrophobic sub-pockets via planar aromatic cores.46
The application of a structure-based pharmacophore model (SBPM), derived from the HIV-1 protease and its crystallized ligand, for virtual screening led to the identification of the virtual hits: Molport-001-545-102, Molport-001-768-271, Molport-002-140-561, Molport-002-853-462, Molport-008-344-770, Molport-012-192-987, Molport-023-246-998, Molport-023-319-072, and Molport-051-594-682. Subsequent, molecular docking studies against 3SPK (PDB ID: HIV-1 protease) revealed extensive hydrophilic and hydrophobic interactions between the top-scoring ligands and the catalytic residues located within the protease binding pocket. Tipranavir, a clinically approved HIV protease inhibitor, was used as a reference ligand. It exhibited characteristic hydrogen bonding and hydrophobic interactions with the key active site residues Asp25, Asp30, Asp29, and Ile50, validating the reliability of the docking workflow and serving as a structural benchmark for comparative evaluation. Among the screened virtual hits, Molport-008-344-770, Molport-023-246-998, and Molport-001-545-102 displayed the most favorable binding orientations and docking energies within the catalytic pocket. Two-dimensional (2D) interaction mapping of these ligand–protein complexes revealed that all three hits formed multiple hydrophilic and hydrophobic contacts with amino acid residues Asn25, Asp29, Asp30, Gly48, Gly49, Ile50, Leu23, Pro81, Ala28, and Thr82—the key residues critical for proteolytic function. Hydrophilic interactions were primarily mediated by amide, carbonyl, and sulphonamide groups, which acted as hydrogen bond donors and acceptors, establishing strong polar contacts with residues such as Asn25, Asp29, and Asp30. The π–π stacking and van der Waals interactions observed between aromatic scaffolds of the ligands and residues Thr82, Pro81, and Leu23 contributed to nonpolar stabilization and overall binding complementarity within the protease groove. The coexistence of both hydrogen bonding and hydrophobic contacts provided optimal structural anchoring and conformational stabilization of the ligands within the binding site. Among the three top hits, Molport-008-344-770 exhibited the strongest binding affinity with a docking energy of −135.824 kcal/mol and the lowest minimized root mean square deviation (mRMSD = 1.335 Å). The ligand established dual hydrogen bonds with Asn25A and Asn25B, a hallmark interaction observed in potent HIV protease inhibitors, and extensive hydrophobic interactions with Thr82, Leu23, Pro81, and Ala28. The naphthol–aryl conjugated ring system within this molecule enhanced π–π stacking and lipophilic contacts, promoting tight binding and minimal conformational drift. The compound’s balanced lipophilicity (iLogP = 2.99) and high gastrointestinal absorption further support its potential for oral bioavailability. Molport-023-246-998, with a docking energy of −127.644 kcal/mol and an mRMSD of 1.851 Å, demonstrated a complementary binding profile driven by its sulphonamide and phenolic functional groups, which formed strong hydrogen bonds with Asp30 and Ile50. The carboxyl and hydroxyl moieties increased the compound’s polarity (TPSA = 161.41 Ų) and solubility (Log S = −2.39), making it the most soluble among the three hits. Although its large polar surface area contributed to low GI absorption, the high density of hydrogen bond donors (nHBD = 5) facilitated robust polar interactions within the protease active site, suggesting strong potential for aqueous-phase binding stability. Molport-001-545-102 also showed excellent docking results with an energy of −127.644 kcal/mol and mRMSD = 1.367 Å, forming multiple hydrogen bonds with Ile50, Asn25, Gly48, and Asp29, as well as π–cation and hydrophobic interactions with Arg8 and Leu23. The sulphonamide group in this compound acted as a key polar anchor, mimicking the carboxylate interaction motif observed in Tipranavir, while the cyclohexane carboxylic acid substituent introduced conformational flexibility and contributed to hydrophobic pocket accommodation. With moderate lipophilicity (iLogP = 2.14) and high GI absorption, Molport-001-545-102 demonstrated a well-balanced pharmacokinetic and binding profile indicative of lead-like potential. Structurally, all three hits share common pharmacophoric features, including amide and sulphonamide linkages, planar aromatic rings, and polarity-distributed substituents, which collectively support high-affinity, non-covalent interactions with HIV-1 protease residues. These features mimic the peptide-bond recognition mechanism exploited by known protease inhibitors, thereby facilitating selective and stable occupation of the enzyme’s active cleft. The aromatic ring systems of the hits enable π–π stacking, while the hydrophilic terminal groups enhance hydrogen bonding and aqueous solubility two essential properties for effective protease inhibition and systemic bioavailability (Table 5 and Figure 4). The SwissADME-predicted physicochemical and pharmacokinetic parameters confirmed favorable drug-like characteristics for all three compounds. None of the hits violated Lipinski’s rule of five, and all demonstrated manageable synthetic accessibility (SA = 2.22–3.62), suggesting practical feasibility for laboratory synthesis. Molport-008-344-770 and Molport-001-545-102 exhibited high GI absorption, while Molport-023-246-998, though more polar, achieved superior solubility (Log S = −2.39). All three were predicted to be non–blood–brain barrier permeant, aligning with the desired pharmacokinetic behavior of HIV protease inhibitors that act peripherally rather than centrally. Overall, Molport-008-344-770 emerged as the most promising hit, displaying the strongest binding energy, high structural complementarity, and optimal physicochemical balance. Molport-023-246-998 demonstrated excellent solubility and a high degree of polar interaction, positioning it as a potential candidate for formulation optimization. Molport-001-545-102, with its extensive hydrogen bonding and hydrophobic residue coverage, offers a valuable scaffold for further structural refinement aimed at potency enhancement. Collectively, the consistent presence of sulphonamide, amide, and aromatic frameworks across these three hits supports their potential as lead-like scaffolds for next-generation HIV-1 protease inhibitor development. Our results fit well with current structural and computational understanding of HIV-1 protease inhibition. Crystal and structural analyses show Tipranavir, and other clinical inhibitors stabilize the active site and flap region via dual hydrogen bonds to the catalytic Asp25/Asp25′ dyad and extensive hydrophobic/aromatic contacts with residues such as Ile50, Leu23 and the flap glycine residues.47
The application of a structure-based pharmacophore model (SBPM), derived from the Dengue viral protease and a bioactive ligand, for virtual screening led to the identification of the virtual hits: Molport-000-808-297, Molport-001-740-947, Molport-002-723-211, Molport-035-706-134, Molport-039-138-765, Molport-039-338-798, Molport-044-726-090, Molport-044-727-269, Molport-044-727-279, and Molport-044-727-351. Subsequent, molecular docking studies against dengue virus protease (PDB ID: 2FOM) revealed a consistent pattern of hydrophilic and hydrophobic interactions between the selected ligands and key amino acid residues within the catalytic binding pocket. Anthracene-1,2,7,9-tetraol, a known inhibitor, served as the reference ligand and exhibited hydrogen bonding with Ser135 and Gly151, as well as hydrophobic contact with Gly153. This validated the docking protocol and provided a structural benchmark for evaluating the affinity and interaction profiles of the virtual hits. Among the screened compounds, Molport-044-727-279, Molport-000-808-297, and Molport-001-740-947 displayed the most favorable binding orientations and lowest binding energies, suggesting strong potential as DENV protease inhibitors. Two-dimensional (2D) interaction visualizations indicated that the top three hits established multiple hydrophilic contacts with residues Gly151, Gly153, Ser135, and Tyr161, alongside hydrophobic interactions involving His51, Leu128, Pro132, and Tyr150. Hydrogen bonding was primarily mediated by hydroxyl and carbonyl groups on the ligands interacting with backbone amide or carbonyl groups of Gly151 and Gly153. π–π stacking interactions were observed between aromatic moieties of the ligands and the imidazole or phenolic side chains of His51 and Tyr161, respectively, reinforcing non-covalent stabilization within the protease active site. The hydrophobic stabilization was contributed by fused aromatic and alkyl moieties engaging Leu128 and Pro132, both of which form part of the enzyme’s hydrophobic pocket adjacent to the catalytic triad. Among the hits, Molport-044-727-279 demonstrated the most favorable binding energy (−121.042 kcal/mol) and optimal spatial complementarity with the active-site residues. This ligand formed dual hydrogen bonds with Gly151 and Gly153, π–π stacking with Tyr161, and hydrophobic contacts with His51, Leu128, and Pro132. The presence of a sugar-like cyclic scaffold with multiple hydroxyl substituents enabled directional hydrogen bonding, while the conjugated aromatic system enhanced π–π stabilization and hydrophobic packing within the catalytic pocket. Molport-000-808-297 showed comparable affinity (−118.633 kcal/mol) and a strong network of interactions involving hydrogen bonds with Gly151 and Gly153, and hydrophobic contacts with Tyr150, Pro132, His51, and Leu128. The presence of dichlorophenyl and hydroxyl-substituted rings provided both lipophilic and polar features that favored balanced binding. Molport-001-740-947 exhibited slightly weaker but still significant binding energy (−116.903 kcal/mol), characterized by π–π stacking with Tyr161 and hydrogen bonding with Gly153 and Ser135. The presence of multiple hydroxyl and methoxy functionalities facilitated solvation potential and stable anchoring through hydrogen bonding. Structurally, all three hits shared a common polyhydroxylated heterocyclic scaffold reminiscent of tetrahydrofuran or sugar-like ring systems, consistent with their strong hydrophilic interaction profiles. These scaffolds provided multiple hydrogen-bond donors and acceptors that mimic peptide substrate recognition within the catalytic cleft of DENV protease. The attached aromatic rings in the hits further contributed to π–π stacking with aromatic residues and hydrophobic packing near His51 and Tyr161, both of which play critical roles in stabilizing substrate–enzyme complexes. The observed interaction geometries suggest that these ligands effectively occupy the protease’s active pocket, engaging residues essential for catalytic activity, thereby indicating their potential as non-covalent, reversible inhibitors (Table 6 and Figure 5). SwissADME-based in silico profiling revealed that all three hits exhibited drug-like properties within acceptable physicochemical limits. None of the top compounds violated more than one of Lipinski’s rule-of-five criteria, and all showed logP values between 1.56 and 1.84, reflecting balanced lipophilicity conducive to oral absorption. The topological polar surface areas (TPSA) ranged from 166.89 to 187.12 Ų, indicating moderately high polarity consistent with the multiple hydroxyl groups observed in their structures. Solubility analysis showed all three ligands to be soluble, with Log S (ESOL) values between −3.55 and −3.87, comparable to the reference inhibitor (−3.99). Gastrointestinal absorption was predicted to be low for the top three hits, which is expected due to their relatively high molecular weights (430–474 g/mol) and polar surface areas. All compounds were predicted to be non–blood–brain barrier (BBB) permeant, an advantageous property for peripheral viral protease inhibitors, reducing the risk of CNS-related adverse effects. Synthetic accessibility (SA) values ranged between 4.58 and 5.06, reflecting moderate synthetic feasibility for early-stage medicinal chemistry optimization. When compared to the reference inhibitor Anthracene-1,2,7,9-tetraol (binding energy: −90.1099 kcal/mol), all three hits demonstrated significantly higher binding affinities and more extensive hydrogen bonding and π–π stacking networks. The hydrophilic–hydrophobic balance observed in these ligands suggests enhanced stabilization within the protease active site through both polar and non-polar interactions. Among the three, Molport-044-727-279 emerged as the most promising hit, combining the lowest binding energy, favorable interaction geometry, and strong hydrogen-bonding with key catalytic residues (Gly151 and Gly153). Molport-000-808-297 followed closely with a balanced interaction profile and favorable hydrophobic packing. Molport-001-740-947, while slightly less potent, displayed excellent polar complementarity and π–π stacking potential, representing a valuable scaffold for structural modification. Overall, the consistent involvement of residues Gly151, Gly153, His51, Tyr161, and Ser135 across the hits confirms the essential binding motif and pharmacophoric pattern required for DENV protease inhibition. The combination of aromatic scaffolds, hydroxyl substituents, and sugar-like frameworks highlights their potential as lead candidates for further optimization in the design of potent, selective dengue virus protease inhibitors. Our results are consistent with current structural and computational knowledge of dengue NS2B–NS3 protease inhibition.39 The catalytic and substrate-binding pockets observed in the 2FOM crystal structure explain why residues Gly151/Gly153, Ser135 and His51 frequently form the backbone of ligand interactions; see the deposited structure and 3D view.48 Recent in-silico studies summarise that potent NS2B–NS3 inhibitors commonly combine polyhydroxylated or polar scaffolds (for directional H-bonding to Gly151/Gly153/Ser135) with fused/aromatic systems (for π–π and hydrophobic packing near His51, Tyr150/Tyr161 and Leu128), features which were matched to the patterns identified in our study.49
The application of a structure-based pharmacophore model (SBPM), derived from the ZIKV protease and a bioactive ligand, for virtual screening led to the identification of the virtual hits: Molport-000-743-059, Molport-000-998-436, Molport-002-603-479, Molport-002-888-347, Molport-046-596-283, Molport-046-703-191, ZINC000004825072, ZINC000005487055, ZINC000015953149, and ZINC000019881412. Subsequent, molecular docking studies against Zika virus protease (PDB ID: 5H4I) revealed a consistent pattern of hydrophilic and hydrophobic interactions between the selected ligands and key amino acid residues within the catalytic binding pocket. N-(5-nitro-6-(piperazin-1-yl)pyridin-2-yl)-1H-indazol-5-amine, a known inhibitor, served as the reference ligand and exhibited hydrogen bonding with Tyr150 and hydrophobic interactions with Ala132 and Tyr161. The indazole ring established π–π stacking with the aromatic side chain of Tyr161, validating the docking protocol and providing a structural benchmark for assessing the virtual screening hits. Among the screened compounds, Molport-002-888-347, ZINC00004825072, and ZINC000015953149 displayed the most favorable binding orientations and lowest docking energies, suggesting strong potential as ZIKV protease inhibitors. The docking energies of these top three hits were −117.7430, −118.4590, and −122.6440 kcal/mol, respectively—each notably lower than that of the reference inhibitor (−99.415 kcal/mol)indicating greater binding affinity and conformational stability within the protease catalytic site. Two-dimensional (2D) interaction analysis revealed that the top three ligands established multiple hydrophilic interactions with residues Gly151, Gly153, Asp75, Asp83, Asp129, and Tyr161, in addition to hydrophobic contacts involving His51, Ala132, and Tyr150. Hydrogen bonding was primarily mediated by hydroxyl, carbonyl, and nitro functional groups, while π–π stacking interactions were observed between aromatic moieties of the ligands and the imidazole or phenolic rings of His51 and Tyr161, respectively, reinforcing non-covalent stabilization within the catalytic pocket. The hydrophobic stabilization was further contributed by the aromatic or fused ring systems of the ligands engaging Ala132 and Tyr161, both of which form part of the hydrophobic subsite adjacent to the protease’s catalytic triad. Among the three hits, ZINC000015953149 demonstrated the most favorable binding energy (−122.6440 kcal/mol) and optimal spatial complementarity with catalytic residues Asp83, His51, and Tyr161. This ligand exhibited hydrogen bonding with Asp83, π–π stacking with His51, and hydrophobic contacts with Ala132 and Tyr161. Structurally, ZINC000015953149 possesses a conjugated electrophilic core with a thioamide, nitroaromatic, and α,β-unsaturated system, suggesting potential covalent character through Michael acceptor-like reactivity. The presence of nitro groups enhances electrophilicity and contributes to ionic interactions with negatively charged residues within the protease active site. ZINC00004825072, with a nucleoside-like scaffold comprising a purine base and sugar ring, exhibited strong hydrogen bonding with Tyr161 and hydrophobic interactions with His51. Its diazo (-N₃) functionality potentially enhances electrophilic activity and contributes to stable anchoring within the enzyme’s polar pocket. Molport-002-888-347, a chromone-like molecule with nitroaromatic and chloro substituents, showed hydrophobic contacts with His51, Tyr161, and Ala132, supporting favorable π–π stacking and lipophilic complementarity within the binding site. The hydroxymethyl and carbonyl substituents of this compound further contributed to directional hydrogen bonding and stabilization. Structurally, all three hits shared the presence of aromatic and heteroaromatic ring systems responsible for π–π stacking and hydrophobic stabilization. The nitro, diazo, and hydroxyl substituents contributed to the formation of hydrogen bonds and potential ionic bridges with aspartate residues within the active pocket, thereby enhancing overall affinity. The coexistence of polar and nonpolar functionalities indicates a favorable hydrophilic–hydrophobic balance crucial for protease inhibition (Table 7 and Figure 6). SwissADME-based in silico profiling revealed that all three hits displayed acceptable physicochemical and pharmacokinetic properties consistent with early-stage drug candidates. None of the compounds violated more than one of Lipinski’s rule-of-five criteria. The calculated lipophilicity (iLogP) values ranged between −0.90 and 1.68, suggesting a balance between aqueous solubility and membrane permeability. Topological polar surface areas (TPSA) ranged from 134.28 to 157.81 Ų, reflecting high polarity consistent with their multiple hydrogen bond donors and acceptors. Solubility analysis showed that ZINC00004825072 was very soluble (Log S = −2.00), Molport-002-888-347 was soluble (−3.51), and ZINC000015953149 moderately soluble (−4.93). All three compounds were predicted to exhibit low gastrointestinal absorption and were non–blood–brain barrier (BBB) permeant, a desirable property minimizing CNS exposure for antiviral therapeutics. Synthetic accessibility (SA) values ranged from 3.61 to 4.34, indicating moderate ease of synthesis and favorable prospects for medicinal chemistry optimization. When compared to the reference inhibitor N-(5-nitro-6-(piperazin-1-yl)pyridin-2-yl)-1H-indazol-5-amine, all three hits demonstrated superior binding energies and a more extensive hydrogen bonding and π–π stacking network involving key residues His51, Asp83, Ala132, and Tyr161. The shared presence of aromatic frameworks and polar substituents allowed strong complementarity to the active site topology, stabilizing both the catalytic cleft and substrate-recognition motifs. Overall, ZINC000015953149 emerged as the most promising hit due to its lowest binding energy, extensive interaction network, and electrophilic pharmacophore, followed closely by ZINC00004825072 and Molport-002-888-347. The consistent involvement of residues His51, Asp83, Ala132, and Tyr161 across all three ligands highlights the essential pharmacophoric motif required for ZIKV protease inhibition. The combined presence of nitroaromatic, diazo, and heterocyclic scaffolds underscores their potential as robust lead candidates for further optimization toward the development of selective, non-covalent ZIKV protease inhibitors. Our computational results are well supported by recent structural and discovery work on Zika NS2B–NS3 protease: the 5H4I crystal coordinates used as your docking template define the catalytic cleft and confirm the relevance of residues such as His51, Asp75, Ala132 and Tyr161 for ligand engagement.50 In addition, structure-based analyses and experimental inhibitor studies have consistently reported hydrogen-bond interactions with Gly151/Gly153 and directional contacts involving Tyr161 and Ala132 in validated hits, further reinforcing the relevance of these interaction patterns.51
Conclusion
The present study demonstrated that virtual hits identified through structure-based pharmacophore modeling (SBPM) exhibit in silico stable binding profiles at the protease target sites of multiple viral pathogens. Five viral protease targets corresponding to SARS-CoV-2, Hepatitis C virus, Human Immunodeficiency Virus, Dengue virus, and Zika virus were selected from the RCSB Protein Data Bank. Individual pharmacophore models were generated for each target and utilized for virtual screening. Nine virtual hits were identified for the HIV protease (PDB ID: 3SPK), while ten hits each were identified for the ZIKV (5H4I), SARS-CoV-2 (8HUR), DENV (2FOM), and HCV (4WF8) proteases. Compounds retrieved from the Molport and ZINC databases via the Pharmit web tool were further validated through molecular docking using iGEMDOCK, which provided binding energy estimations. The SWISSADME tool was employed to evaluate physicochemical properties, and ProteinPlus (PoseEdit function) was used to visualize 2D ligand–protein interactions. The results confirmed that all virtual hits displayed stable and favourable binding interactions within the target active sites, consistent with or exceeding those of reference inhibitors. The identified chemical scaffolds and structural features responsible for binding stability were also characterized. Overall, this investigation establishes a systematic and computationally efficient framework for the discovery of antiviral protease inhibitors. While in silico approaches significantly reduce time and cost in early-stage drug discovery by screening large compound libraries, experimental validation through in vitro assays, synthesis of hit compounds, and subsequent lead optimization will be essential for confirming biological activity and improving pharmacological properties. Scaffold optimization in future studies will further enhance binding affinity and minimize potential side effects, advancing these virtual hits toward viable antiviral drug candidates.
Acknowledgement
All authors express their sincere gratitude to the Vice-Chancellor of IMU University, the Dean of the School of Pharmacy, the Dean of the School of Postgraduate Studies, and the Director and Deputy Directors of the Institute for Research, Development & Innovation (IRDI) for providing the facilities necessary to complete this research project, undertaken as part of the fulfilment of the Master of Science (MSc) in Analytical and Pharmaceutical Chemistry programme at IMU University.
Funding Source
This research was funded by the IMU University Joint Committee on Research and Ethics under the approved project, Project ID: MSc in Analytical and Pharmaceutical Chemistry-MAPC I-2024 (03).
Conflict of Interest
The author(s) do not have any conflict of interest.
Data availability
This statement does not apply to this article
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable
Author Contributions
- Vasudeva Rao Avupati: Conceptualization, Project Administration, Funding Acquisition, Resources, Supervision, Review & Editing.
- Fabian Mok Ping Xiang: Methodology, Visualization, Data Collection, Analysis, Writing.
References
- Giordano D, Biancaniello C, Argenio MA, Facchiano A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals. 2022;15(5). doi:10.3390/ph15050646
CrossRef - Adamson CS, Chibale K, Goss RJM, Jaspars M, Newman DJ, Dorrington RA. Antiviral drug discovery: Preparing for the next pandemic. Chem Soc Rev. 2021;50(6). doi:10.1039/d0cs01118e
CrossRef - Rietdijk J, Tampere M, Pettke A, et al. A phenomics approach for antiviral drug discovery. BMC Biol. 2021;19(1). doi:10.1186/s12915-021-01086-1
CrossRef - Sosnina EA, Sosnin S, Nikitina AA, Nazarov I, Osolodkin DI, Fedorov M V. Recommender Systems in Antiviral Drug Discovery. ACS Omega. 2020;5(25). doi:10.1021/acsomega.0c00857
CrossRef - Kaparapu J, Krishna Prasad M, Mohan Narasimha Rao G. Antiviral potentials of marine algal bioactive compounds for coronavirus drug discovery. In: Coronavirus Drug Discovery: Volume 2: Antiviral Agents from Natural Products and Nanotechnological Applications. Vol 2. 2022. doi:10.1016/B978-0-323-95574-4.00002-0
CrossRef - Dong J, Varbanov M, Philippot S, Vreken F, Zeng W bin, Blay V. Ligand-based discovery of coronavirus main protease inhibitors using MACAW molecular embeddings. J Enzyme Inhib Med Chem. 2023;38(1). doi:10.1080/14756366.2022.2132486
CrossRef - Bafna K, Krug RM, Montelione GT. Structural Similarity of SARS-CoV2 Mpro and HCV NS3/4A Proteases Suggests New Approaches for Identifying Existing Drugs Useful as COVID-19 Therapeutics. ChemRxiv. Published online 2020. doi:10.26434/chemrxiv.12153615
CrossRef - Wamser R, Pach S, Arkona C, et al. A Critical Study on Acylating and Covalent Reversible Fragment Inhibitors of SARS-CoV-2 Main Protease Targeting the S1 Site with Pyridine. ChemMedChem. 2023;18(9). doi:10.1002/cmdc.202200635
CrossRef - Talele TT, Arora P, Kulkarni SS, et al. Structure-based virtual screening, synthesis and SAR of novel inhibitors of hepatitis C virus NS5B polymerase. Bioorg Med Chem. 2010;18(13):4630-4638. doi:10.1016/j.bmc.2010.05.030
CrossRef - Ng TI, Tripathi R, Reisch T, et al. In vitro antiviral activity and resistance profile of the next-generation hepatitis C virus NS3/4A protease inhibitor glecaprevir. Antimicrob Agents Chemother. 2018;62(1). doi:10.1128/AAC.01620-17
CrossRef - Vicenti I, Rosi A, Saladini F, et al. Naturally occurring hepatitis C virus (HCV) NS3/4A protease inhibitor resistance-related mutations in HCV genotype 1-infected subjects in Italy. Journal of Antimicrobial Chemotherapy. 2012;67(4). doi:10.1093/jac/dkr581
CrossRef - Patel BA, Krishnan R, Khadtare N, et al. Design and synthesis of l- and d-phenylalanine derived rhodanines with novel C5-arylidenes as inhibitors of HCV NS5B polymerase. Bioorg Med Chem. 2013;21(11):3262-3271. doi:10.1016/j.bmc.2013.03.041
CrossRef - Debouck C. The HIV-1 Protease as a Therapeutic Target for AIDS. AIDS Res Hum Retroviruses. 1992;8(2). doi:10.1089/aid.1992.8.153
CrossRef - Monica G La, Lauria A, Bono A, Martorana A. Off-target-based design of selective hiv-1 protease inhibitors. Int J Mol Sci. 2021;22(11). doi:10.3390/ijms22116070
CrossRef - Ali A, Bandaranayake RM, Cai Y, et al. Molecular basis for drug resistance in HIV-1 protease. Viruses. 2010;2(11). doi:10.3390/v2112509
CrossRef - Arrigoni R, Santacroce L, Ballini A, Palese LL. AI-Aided Search for New HIV-1 Protease Ligands. Biomolecules. 2023;13(5). doi:10.3390/biom13050858
CrossRef - Okafor SN, Meyer A, Gadsden J, et al. Drug Reprofiling to Identify Potential HIV-1 Protease Inhibitors. Molecules. 2023;28(17). doi:10.3390/molecules28176330
CrossRef - Heal JW, Jimenez-Roldan JE, Wells SA, Freedman RB, Römer RA. Inhibition of HIV-1 protease: The rigidity perspective. Bioinformatics. 2012;28(3). doi:10.1093/bioinformatics/btr683
CrossRef - Wu H, Bock S, Snitko M, et al. Novel dengue virus NS2B/NS3 protease inhibitors. Antimicrob Agents Chemother. 2015;59(2). doi:10.1128/AAC.03543-14
CrossRef - Lin KH, Ali A, Rusere L, Soumana DI, Kurt Yilmaz N, Schiffer CA. Dengue Virus NS2B/NS3 Protease Inhibitors Exploiting the Prime Side. J Virol. 2017;91(10). doi:10.1128/jvi.00045-17
CrossRef - Osman H, Idris NH, Kamarulzaman EE, Wahab HA, Hassan MZ. 3,5-Bis(arylidene)-4-piperidones as potential dengue protease inhibitors. Acta Pharm Sin B. 2017;7(4). doi:10.1016/j.apsb.2017.04.009
CrossRef - Tomlinson SM, Watowich SJ. Anthracene-based inhibitors of dengue virus NS2B-NS3 protease. Antiviral Res. 2011;89(2). doi:10.1016/j.antiviral.2010.12.006
CrossRef - Li H, Zhu L, Hou S, Yang J, Wang J, Liu J. An inhibition model of BPTI to unlinked dengue virus NS2B-NS3 protease. FEBS Lett. 2014;588(17). doi:10.1016/j.febslet.2014.05.063
CrossRef - Saleem HN, Kousar S, Jiskani AH, Sohail I, Faisal A, Saeed M. Repurposing of investigational cancer drugs: Early phase discovery of dengue virus NS2B/NS3 protease inhibitors. Arch Pharm (Weinheim). 2023;356(11). doi:10.1002/ardp.202300292
CrossRef - Hill ME, Yildiz M, Hardy JA. Cysteine Disulfide Traps Reveal Distinct Conformational Ensembles in Dengue Virus NS2B-NS3 Protease. Biochemistry. 2019;58(6). doi:10.1021/acs.biochem.8b00978
CrossRef - Bharadwaj S, Lee KE, Dwivedi VD, et al. Discovery of Ganoderma lucidum triterpenoids as potential inhibitors against Dengue virus NS2B-NS3 protease. Sci Rep. 2019;9(1). doi:10.1038/s41598-019-55723-5
CrossRef - Nitsche C, Schreier VN, Behnam MAM, Kumar A, Bartenschlager R, Klein CD. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J Med Chem. 2013;56(21):8389-8403. doi:10.1021/jm400828u
CrossRef - Kumar A, Liang B, Aarthy M, et al. Hydroxychloroquine Inhibits Zika Virus NS2B-NS3 Protease. ACS Omega. 2018;3(12). doi:10.1021/acsomega.8b01002
CrossRef - Gruba N, Rodriguez Martinez JI, Grzywa R, et al. Substrate profiling of Zika virus NS2B-NS3 protease. FEBS Lett. 2016;590(20). doi:10.1002/1873-3468.12443
CrossRef - Chen X, Yang K, Wu C, et al. Mechanisms of activation and inhibition of Zika virus NS2B-NS3 protease. Cell Res. 2016;26(11). doi:10.1038/cr.2016.116
CrossRef - Quek JP, Liu S, Zhang Z, et al. Identification and structural characterization of small molecule fragments targeting Zika virus NS2B-NS3 protease. Antiviral Res. 2020;175. doi:10.1016/j.antiviral.2020.104707
CrossRef - Lei J, Hansen G, Nitsche C, Klein CD, Zhang L, Hilgenfeld R. Crystal structure of zika virus ns2b-ns3 protease in complex with a boronate inhibitor. Science (1979). 2016;353(6298). doi:10.1126/science.aag2419
CrossRef - Li Y, Zhang Z, Phoo WW, et al. Structural Dynamics of Zika Virus NS2B-NS3 Protease Binding to Dipeptide Inhibitors. Structure. 2017;25(8). doi:10.1016/j.str.2017.06.006
CrossRef - Phoo WW, Zhang Z, Wirawan M, et al. Structures of Zika virus NS2B-NS3 protease in complex with peptidomimetic inhibitors. Antiviral Res. 2018;160. doi:10.1016/j.antiviral.2018.10.006
CrossRef - Hammerschmidt SJ, Huber S, Braun NJ, Lander M, Steinmetzer T, Kersten C. Thermodynamic characterization of a macrocyclic Zika virus NS2B/NS3 protease inhibitor and its acyclic analogs. Arch Pharm (Weinheim). 2023;356(4). doi:10.1002/ardp.202200518
CrossRef - Lin C, Jiang H, Li W, et al. Structural basis for the inhibition of coronaviral main proteases by ensitrelvir. Structure. 2023;31(9). doi:10.1016/j.str.2023.06.010
CrossRef - Soumana DI, Ali A, Schiffer CA. Structural analysis of asunaprevir resistance in HCV NS3/4A protease. ACS PublicationsDI Soumana, A Ali, CA SchifferACS chemical biology, 2014•ACS Publications. 2014;9(11):2485-2490. doi:10.1021/CB5006118
CrossRef - Wang Y, Liu Z, Brunzelle JS, et al. The higher barrier of darunavir and tipranavir resistance for HIV-1 protease. Biochem Biophys Res Commun. 2011;412(4). doi:10.1016/j.bbrc.2011.08.045
CrossRef - Erbel P, Schiering N, D’Arcy A, et al. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat Struct Mol Biol. 2006;13(4). doi:10.1038/nsmb1073
CrossRef - Zhang Z, Li Y, Loh YR, et al. crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science (1979). 2016;354(6319). doi:10.1126/science.aai9309
CrossRef - Shin HJ, Kim MH, Lee JY, et al. Structure-based virtual screening: Identification of a novel NS2B-NS3 protease inhibitor with potent antiviral activity against zika and dengue viruses. mdpi.com. https://www.mdpi.com/2076-2607/9/3/545
CrossRef - Gevorgyan S, Khachatryan H, Shavina A, Gharaghani S, Zakaryan H. Targeting SARS-CoV-2 main protease: a comprehensive approach using advanced virtual screening, molecular dynamics, and in vitro validation. SpringerS Gevorgyan, H Khachatryan, A Shavina, S Gharaghani, H ZakaryanVirology Journal, 2024•Springer. 2024;21(1):330. doi:10.1186/S12985-024-02607-4
CrossRef - Gao S, Song L, Ye B, et al. Miniaturized click chemistry and direct screening facilitate the discovery of triazole piperazine SARS-CoV-2 M pro inhibitors with improved metabolic stability. pubs.rsc.orgS Gao, L Song, B Ye, M Yang, J Li, M Gu, AE Tollefson, K Toth, P Zhan, X LiuRSC Medicinal Chemistry, 2025•pubs.rsc.org. Accessed November 19, 2025. https://pubs.rsc.org/en/content/articlehtml/2025/md/d4md00555d
CrossRef - Alagawani I, Sciences FWA, 2025 undefined. In Silico Development of SARS-CoV-2 Non-Covalent Mpro Inhibitors: A Review. mdpi.com. Accessed November 19, 2025. https://www.mdpi.com/2076-3417/15/12/6544
CrossRef - Iman K, Mirza MU, Sadia F, Froeyen M, Trant JF, Chaudhary SU. Pharmacophore-Assisted Covalent Docking Identifies a Potential Covalent Inhibitor for Drug-Resistant Genotype 3 Variants of Hepatitis C Viral NS3/4A Serine Protease. Viruses. 2024;16(8). doi:10.3390/V16081250
CrossRef - Qu M, Gao M, Sang X, et al. Structural bioinformatics approaches for predicting novel drug targets in hepatitis C virus proteins: a comprehensive analysis. nature.comM Qu, M Gao, X Sang, M Yu, Z Guan, W ChangScientific Reports, 2025•nature.com. doi:10.1038/s41598-025-12563-w
CrossRef - Muzammil S, Armstrong AA, Kang LW, et al. Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations. journals.asm.orgS Muzammil, AA Armstrong, LW Kang, A Jakalian, PR Bonneau, V Schmelmer, LM AmzelJournal of virology, 2007•journals.asm.org. 2007;81(10):5144-5154. doi:10.1128/JVI.02706-06
CrossRef - Bharadwaj S, Lee KE, Dhar Dwivedi V, et al. Discovery of Ganoderma lucidum triterpenoids as potential inhibitors against Dengue virus NS2B-NS3 protease. nature.com. (7). doi:10.1038/s41598-019-55723-5
CrossRef - Norshidah H, Herng Leow C, Ezatul Ezleen K, et al. Assessing the potential of NS2B/NS3 protease inhibitors biomarker in curbing dengue virus infections: In silico vs. In vitro approach. frontiersin.orgH Norshidah, CH Leow, KE Ezleen, HA Wahab, R Vignesh, A Rasul, NS LaiFrontiers in cellular and infection microbiology, 2023•frontiersin.org. 2023;13. doi:10.3389/FCIMB.2023.1061937/FULL
CrossRef - Zhang Z, Li Y, Loh YR, et al. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. science.org. 2016;354(6319):1597-1600. doi:10.1126/SCIENCE.AAI9309
CrossRef - Shin H, Kim M, Lee J, et al. Structure-based virtual screening: Identification of a novel NS2B-NS3 protease inhibitor with potent antiviral activity against zika and dengue viruses. mdpi.com. Accessed November 19, 2025. https://www.mdpi.com/2076-2607/9/3/545
CrossRef

