
Manuscript accepted on :18-11-2025
Published online on: 04-12-2025
Plagiarism Check: Yes
Reviewed by: Dr. M Mohan Varma
Second Review by: Dr. Nadhim M. H
Final Approval by: Dr. Anton R Keslav
Safia Khalil Ali 1,3 and Elshazali Widaa Ali 2,3*
and Elshazali Widaa Ali 2,3*
1Department of Hematology, Faculty of Medical Laboratory Sciences, Omdurman Islamic University, Khartoum, Sudan.
2Department of Medical Laboratory Sciences, College of Applied Medical Sciences, University of Bisha, Bisha, Saudi Arabia.
3Department of Hematology, Faculty of Medical Laboratory Sciences, Al Neelain University, Khartoum, Sudan
Corresponding Author E-mail:elshazali@ub.edu.sa
DOI : https://dx.doi.org/10.13005/bpj/3324
Abstract
β-thalassemia major (βTM) is characterized by severe anemia that necessitates regular transfusions, leading to iron overload. Mutations in the HFE gene have been reported to affect iron homeostasis and may exacerbate iron accumulation in patients with thalassemia. This study aimed to screen for HFE gene C282Y and E277K mutations in Sudanese children with βTM to assess their impact on the severity of iron overload. A total of 76 children with βTM were enrolled in the study. Genomic deoxyribonucleic acid was extracted from peripheral leukocytes and analyzed for the C282Y and E277K mutations in the HFE gene using polymerase chain reaction amplification and Sanger sequencing. Serum ferritin (SF) levels were determined using a biochemical analyzer to assess the severity of iron overload. The C282Y mutation was absent in all participants, whereas the E277K mutation was identified in 2 (2.6%) patients in a heterozygous state. Furthermore, an intron 3 variant (rs807209) was detected in 16 (22.3%) patients within the same amplicon. Although the mean SF level was lower in patients with the E277K mutation and higher in those with the intron 3 variant than in those carrying the wildtype allele, the differences were not statistically significant (P-values = 0.140 and 0.164, respectively). In conclusion, the HFE C282Y mutation was not detected among Sudanese children with βTM. The E277K mutation was rare (2.6%) and was not significantly associated with the severity of iron overload. Similarly, the intron 3 variant (rs807209) was present in a proportion of patients (22.3%) but had no significant impact on the severity of iron overload.
Keywords
β-thalassemia major; C282Y Mutation; E277K mutation; HFE Gene; Iron Overload
Download this article as:| Copy the following to cite this article: Ali S. K, Ali E. W. HFE Gene C282Y and E277K Mutations as Possible Genetic Modulators of Iron Overload Severity in Transfusion-Dependent β-Thalassemia Major. Biomed Pharmacol J 2025;18(4). |
| Copy the following to cite this URL: Ali S. K, Ali E. W. HFE Gene C282Y and E277K Mutations as Possible Genetic Modulators of Iron Overload Severity in Transfusion-Dependent β-Thalassemia Major. Biomed Pharmacol J 2025;18(4). Available from: https://bit.ly/3KBlPGx |
Introduction
β-Thalassemia major (βTM), also known as Cooley’s anemia, is the most severe form of thalassemia, resulting from mutations in both β-globin alleles that lead to severely reduced or absent α-globin chains. The resulting imbalance in globin chains leads to ineffective erythropoiesis and severe hemolytic anemia, necessitating regular blood transfusions to maintain adequate hemoglobin (Hb) levels.1,2
Regular blood transfusions lead to iron overload and life-threatening complications, as excess iron saturates the transferrin-iron transport system, resulting in the presence of non-transferrin-bound iron (NTBI) that circulates in plasma and is subsequently deposited in susceptible cells.3,4,5 Furthermore, NTBI is unstable and can easily be changed from ferric to ferrous form, resulting in the generation of reactive oxygen species, which cause tissue damage through lipid peroxidation and lead to a variety of health problems.6,7,8 Iron overload-related complications (IOCs) include growth retardation and failure or delay of sexual maturation.9 Later, complications may include the involvement of the heart, liver, and endocrine glands.10 Heart failure and arrhythmias, caused by myocardial siderosis, are the most important life-limiting complications of iron overload in patients with βTM.11
Compliance with iron chelating therapy (ICT) can reduce the frequency and severity of IOCs and improve the survival of βTM patients.12 However, ferritin levels above 2500 μg/l are associated with a higher risk of morbidity and mortality, and levels persistently above this value should trigger an intensification of the chelation regimen.13,14 Hereditary hemochromatosis (HH), also referred to as type I hemochromatosis, is caused by mutations in the HFE gene, which is located on chromosome 6 and encodes the HFE-iron-regulatory protein, a key limiting factor of the duodenal iron absorption. This protein plays a crucial role in iron homeostasis by interacting with transferrin receptors and regulating hepcidin production, a hormone that regulates iron absorption and distribution in the body.15,16
The HFE C282Y mutation results from a G-to-A substitution at nucleotide 845 in the HFE gene, leading to the substitution of cysteine with tyrosine at position 282 of the HFE protein. This mutation is the most frequent mutation associated with HH, accounting for approximately 80-90% of cases in populations of European descent.17-19 The pathophysiology of the C282Y mutation involves the disruption of normal HFE protein function, which is crucial for regulating iron homeostasis; it impairs the interaction of HFE protein with transferrin receptors on cell surfaces, thereby modulating hepcidin expression, leading to decreased hepcidin levels and, consequently, increased intestinal iron absorption.17,20
The E277K mutation is a less common genetic alteration associated with HH. It involves a substitution of glutamic acid (E) with lysine (K) at position 277 of the HFE protein. The functional significance of the E277K mutation is still being elucidated. Studies suggest that this mutation may affect the interaction of the HFE protein with transferrin receptors and a2-microglobulin, potentially leading to impaired signaling pathways that regulate iron metabolism.21 Carriers of E277K mutation have been reported to exhibit varying degrees of iron overload, depending on additional genetic and environmental factors. The presence of other mutations in the HFE gene can influence the phenotypic expression of iron overload in individuals with the E277K mutation. Furthermore, lifestyle factors, such as diet and alcohol consumption, may also modulate the severity of iron overload in carriers of this mutation.22,23
Screening for HFE mutations in patients with βTM can help predict the risk of iron overload, enabling early detection and more effective management to address IOCs.13,24
This study aimed to screen for HFE gene C282Y and E277K mutations in Sudanese children with βTM and explore their influence on the severity of iron overload.
Materials and Methods
This is a descriptive cross-sectional study, in which a total of 76 children diagnosed with βTM were recruited, all were treated with regular blood transfusions and monitored at multiple Hospitals in Khartoum state, Sudan. Blood samples were collected from all participants and used to estimate serum ferritin (SF), perform complete blood count (CBC), and conduct molecular analysis.
Hematological and biochemical analysis
CBC was performed immediately after sample collection using an automated hematology analyzer (Mission HA-360, USA), and SF level was measured using an automated analyzer (Cobas e-411) with Elecsys Ferritin kit (Roche Diagnostics International Ltd, Switzerland).
Molecular analysis
DNA extraction
Genomic DNA was isolated from peripheral leucocytes using the “innuPREP Blood DNA extraction kit” (Analytik Jena, Germany) and stored at −20°C for further analysis.
Polymerase Chain Reaction (PCR)
A PCR reaction mixture (23 μL) was prepared for each sample; it consists of 4 μl ready-to-load master mix, 1 μl of each of the forward (5′-GGGTATTTCCTTCCTCCAACC-3′) and reverse (5′-CTCAGGCACTCCTCTCAACC-3′) primers (Intron biotechnology, South Korea), 2 μl genomic DNA, and 15 μl distilled water. The mixture was amplified using a thermal cycler (Biometra TADVANCED, Germany). The thermocycling conditions included initial denaturation at 95°C for 2 minutes, followed by 35 cycles of [95°C for 30 seconds, 63.3°C for 30 seconds, and 72°C for 30 seconds], and a final extension at 72°C for 5 minutes.
Agarose gel electrophoresis
PCR products (4 μL) were separated on an ethidium bromide-stained agarose gel (1.5%) and visualized using a gel documentation system (Biometra BDA compact, Germany). The optimal size of the product (441 bp) was determined by comparing it with a 100 bp DNA ladder.
DNA sequencing
Sanger DNA sequencing was used to screen the amplified fragment for HFE gene C282Y and E277K mutations (BGI-Genomics Company, China).
Data collection and analysis
Patients’ data were collected from the medical records and analyzed using the Statistical Package for Social Sciences (SPSS), version 25. Qualitative data were presented as frequency and percentage, while quantitative data were presented as mean±standard deviation (SD). The association between qualitative variables was tested using Chi-square and Fisher’s exact tests. The means of quantitative variables were compared by an independent two-sample test and ANOVA. Multivariable logistic regression was conducted to evaluate the association between HFE gene mutations and the development of IOCs, while adjusting for potential confounders, including age, transfusion frequency, iron chelation therapy, and sex.
Results
Demographic and clinical data
A total of 76 Sudanese children with βTM were enrolled in this study; 46 (60.5%) were males and 30 (39.5%) were females; 41 (53.9%) were under the age of 7.
The severity of anemia varied significantly among the study participants; over half (55%) had severe anemia (Hb < 7 g/dl), 30% had moderate anemia (Hb 7–10 g/dl), and 15% had mild anemia (Hb > 10 g/dl). Most patients (92.1%) had elevated SF levels, while the remaining 7.9% had values within the normal range. Approximately two-thirds (67.1%) experienced iron IOCs. The most frequent complication was growth retardation (47.1%), followed by hypersplenism (37.3%), liver disease (5.9%), and diabetes mellitus (2.0%). Although 16 patients (21.1%) were at risk of myocardial iron deposition based on their SF levels (≥ 2500 μg/l), none showed clinical signs of cardiac disease.
About half of the patients (53%) were receiving ICT. Almost all patients, except one, were on subcutaneous deferoxamine, while the remaining one was taking oral deferasirox. The participants demonstrated variable adherence to ICT, as indicated by the long mean duration since the last dose (Mean ± SD: 192 ± 230 days).
The comparison of SF levels according to anemia severity showed that the mean SF level was higher in patients with severe anemia than in those with moderate anemia, and in patients with moderate anemia than those with mild anemia, but the difference was not statistically significant (Mean± SD: 2106.6± 1505.9 μg/l, 2091.7± 1443.4 μg/l, and 1656.1 ± 1395.2 μg/l respectively, P-value = 0.652).
IOCs were significantly more prevalent among patients aged seven years or older compared to those younger than seven, and they increased in parallel with anemia severity. The number of blood transfusions received was significantly higher in patients with IOCs compared to those without complications (median: 73 and 20, respectively, P-value= 0.00). ICT was found to have no significant effect on the frequency of IOCs (Table 1). As shown in Figure 1, a statistically significant positive correlation was observed between S. ferritin levels and the time since the last ICT dose (r=0.310, P-value= 0.046).
Table 1: Association between iron overload complications and age group, anemia severity, and iron chelation therapy
| Variable | Iron overload complications | P. value | ||
| Yes | No | |||
| Age group | <7 years | 21 (51.2%) | 20 (48.8%) | 0.007 |
| ≥7 years | 30 (85.7%) | 5 (14.3%) | ||
| Anemia severity | Mild | 3 (27.3%) | 8 (72.7%) | 0.012 |
| Moderate | 16 (69.6%) | 7 (30.4%) | ||
| Severe | 32 (76.2%) | 10 (23.8%) | ||
| Chelating therapy | Yes | 30 (71.4%) | 12 (28.6%) | 0.373 |
| No | 21 (61.8%) | 13 (38.2%) | ||
P-value significant at≤ 0.05
Frequency of HFE gene mutations
The results of DNA sequencing revealed the absence of HFE C282Y mutation in all the study subjects (Figure 2). The E277K mutation was detected in two patients (2.6%) in heterozygous state (Figure 3). Another single-nucleotide variation, intron 3 C>G (rs807209), was identified in the same sequence amplified for detecting the C282Y and E277K mutations; it was found in 16 (22.3%) patients, one in homozygous state and 15 in heterozygous state (Figure 4).
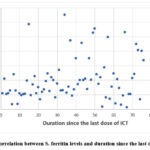 |
Figure 1: Correlation between S. ferritin levels and duration since the last dose of ICT |
 |
Figure 2: Negative C282Y mutation; the alignment shows the wildtype (G) allele in all patients |
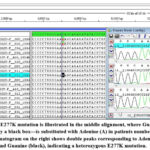 |
Figure 3: The E277K mutation is illustrated in the middle alignment, where Guanine (G)—highlighted by a black box—is substituted with Adenine (A) in patients numbered 11 and 12. |
 |
Figure 4: Intron 3 (rs807209) mutation. The BioEdit alignment shows a nucleotide substitution in which Cytosine (C) is replaced by Guanine (G), demonstrating the base substitution associated with the rs807209 variant. |
Impact of HFE mutations on the severity of iron overload
The two patients with the E277K mutation had lower SF levels than those with the wild-type allele, but the difference was not statistically significant (mean ± SD: 1727.53 ± 1667.6 and 2107.17 ± 1421.9 μg/l, respectively; P-value = 0.140).
Although the only patient with homozygous intron 3 mutation (GG) was found to have a higher SF level (2460 μg/l) compared to those with the heterozygous genotype and those with the wild-type allele, no statistically significant difference was observed (Mean± SD: 1727.53±1667.6 and 2107.17± 1421.9 μg/l, respectively, P-value= 0.164).
Associations of HFE genetic variants (E272K & rs807209) with risk of IOCs
After adjusting for age, transfusion frequency, ICT, and sex, multivariable logistic regression revealed that neither the E272K mutation nor the intron 3 (rs807209) variant was an independent risk factor for IOCs. Age showed a borderline but insignificant positive association, while transfusion frequency was significantly associated with an increased risk of complications. Although not statistically significant, ICT demonstrated a protective trend, and sex had no considerable impact (Table 2).
Table 2: Association of HFE gene E272K and intron 3 rs807209 mutations with risk of IOCs
| Variable | aOR | 95% CI s | P-value* | |
| Lower | Upper | |||
| E272K mutation | 1.39 | 0.003 | 581.47 | 0.915 |
| Intron 3 variant (rs807209) | 0.56 | 0.12 | 2.58 | 0.458 |
| Age | 1.25 | 0.96 | 1.63 | 0.093 |
| Frequency of transfusions | 1.04 | 1.01 | 1.07 | 0.005 |
| Chelation therapy | 0.26 | 0.06 | 1.16 | 0.078 |
| Sex | 1.34 | 0.37 | 4.86 | 0.658 |
*P-value significant at≤ 0.05
Discussion
The HFE protein plays a crucial role in regulating iron metabolism, and mutations in HFE gene account for almost 90% of HH phenotypes in some populations.25,26 This study screened Sudanese children with βTM for the HFE gene C282Y and E277K mutations to explore their effects on iron overload severity. The study included 76 children with βTM receiving regular blood transfusions and monitored at various hospitals in Khartoum state, Sudan.
The results revealed that the majority of children with βTM (92.1%) had high SF ferritin levels, a critical marker of iron overload. Moreover, 21.1% of children had SF levels > 2500 μg/L, indicating an increased risk of myocardial iron loading. This finding is consistent with the established understanding that patients with βTM, who frequently require blood transfusions to manage their severe anemia, are at high risk of iron accumulation due to the absence of a physiological mechanism for iron excretion.7,27 The association between the frequency of IOCs and factors such as the severity of anemia, age group (≥7 years), and the number of blood transfusions received is particularly noteworthy. The severity of anemia in βTM patients often necessitates more frequent transfusions, thereby increasing the iron burden. The relationship between transfusion frequency and iron overload is well-documented in previous studies, with each unit of transfused red blood cells introducing approximately 200-250 mg of iron into the body.28,29 Consequently, patients who receive more transfusions are at an increased risk of developing IOCs.30 The age group is also significant in this context, as children with βTM aged ≥7 years typically accumulate more iron due to the cumulative effects of transfusions over time. This accumulation can lead to a higher incidence of complications, as the organs become increasingly burdened by excess iron. Studies have shown that the risk of developing cardiac complications, for instance, rises significantly in older children and adolescents with βTM, particularly those with high SF levels.31,32 This trend emphasizes the importance of regular monitoring and proactive management of iron levels in this age group to mitigate the risk of long-term complications.
The current study found that ICT did not significantly affect the frequency of IOCs in children with βTM. This finding aligns with the existing literature, which highlights the complexities and limitations of ICT in managing iron overload associated with frequent blood transfusions. Despite the introduction of various iron chelators, the effectiveness of these treatments can vary significantly among patients, and some studies have reported persistent high SF levels even with ICT.7,29 This suggests that while ICT is critical for managing iron overload, it may not be sufficient to prevent complications in all patients. In this study, patients exhibited variable adherence to ICT, as reflected in the long time since the last dose (Mean ± SD: 192.2 ± 230 days). The analysis showed a statistically significant positive correlation between S. ferritin and the time since the previous dose of ICT (r=0.310, P-value= 0.046), which reflects that adherence to ICT is a critical factor influencing its effectiveness. This finding is supported by a previous study, which showed that poor adherence to prescribed chelation regimens can lead to inadequate management of iron levels, resulting in continued complications.32 All patients in this study, except one, were treated with subcutaneous deferoxamine, while a single patient received oral deferasirox. This limited variation in treatment type makes it challenging to evaluate the impact of the type of ICT on the frequency of IOCs.
Researchers have shown that the prevalence of the HFE C282Y mutation varies across different populations. While the mutation is common in European populations, its frequency is significantly lower in non-European groups.33 In the current study, the C282Y mutation was not detected in any of our study participants, indicating no role for this mutation in iron overload among Sudanese children with βTM. This finding is consistent with many studies that reported extreme rarity or even absence of C282Y mutation among βTM patients in some populations.34,35
The E277K mutation was detected in only two patients (2.6%). This finding is consistent with Bradbury et al,36 who reported this substitution as a rare polymorphism in the HFE gene. Karimi et al,37 found E277K mutation in a homozygous state for the first time in Iranian transfused and chelated βTM, but they couldn’t establish or exclude its influence on the endogenous iron loading. Later, Silva et al,21 analyzed the functional consequences of the E277K mutation and reported that it negatively affects both the HFE alternative splicing mechanism and protein interactions and may play a role in the development of HH. Furthermore, E277K substitution was detected in compound heterozygosity with H63D in Portuguese males who presented with altered iron parameters, while it was not found in healthy Portuguese individuals; thus, it was not considered a polymorphism.38
In the present study, statistical insignificance was noted in the mean SF levels between the patients carrying the E277K mutation and those with the wild-type allele.
In the current study, another genetic variant, the intron 3 C>G (rs807209) mutation, was detected by DNA sequencing, with a frequency of 19.7% for the heterozygous state and 1.3% for the homozygous state. Although the only patient with the homozygous mutant genotype (GG) had higher SF levels than those with heterozygous or wild-type genotypes, the difference was statistically insignificant. This mutation was previously reported in the Brazilian population by Campos et al.39 However, to our knowledge, there is no published data on this mutation and iron overload in thalassemic patients. According to data from the ClinVar database (Accession: VCV001292203.4), this variant is classified as a “benign” single-nucleotide variant (SNV). However, this classification is based on a single clinical study,40 and its functional significance remains uncertain. Therefore, future studies using in silico prediction tools or functional assays are recommended to clarify its potential regulatory role.
After controlling for age, frequency of blood transfusion, ICT, and sex, multivariable logistic regression showed that neither the E272K mutation nor the intron 3 (rs807209) variant was an independent risk factor for the development of IOCs. Age showed a borderline insignificant positive association, while transfusion frequency was significantly associated with an increased risk of complications. ICT demonstrated a protective trend, but it was not statistically significant, and sex did not have a considerable effect on the development of IOCs. However, the low frequency of the E277K mutation and the intron 3 rs807209 variant limits the statistical power to detect meaningful associations with iron overload severity or complications.
Conclusion
C282Y mutation of the HFE gene was not reported in Sudanese children with βTM. The E277K mutation was present, but at a low frequency, leaving its role in the severity of iron overload uncertain. The Intron 3 C>G (rs807209) variant of the HFE gene was also identified in our study population. Patients who were homozygous for this mutation had higher SF levels compared to those with the heterozygous state or the wild-type allele, but the difference was not statistically significant. The frequency of blood transfusion was significantly associated with the risk of developing IOCs, while no significant effect was observed for age, sex, or ICT.
Acknowledgment
The authors extend their sincere gratitude to the Faculty of Medical Laboratory Sciences at Omdurman Islamic University and Al Neelain University for their valuable support throughout the study.
Funding Source
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data availability
The manuscript incorporates all datasets produced throughout this research study. HFE gene sequence data have been submitted to NCBI GenBank, and accession numbers have been released (BankIt2525005: OL688473-OL688542).
Ethics Statement
The study was approved by the ethical committee of the Ministry of Health, Khartoum state, Sudan.
Informed consent statement
Informed consent was obtained from the children’s parents before sample collection. All methods were performed in accordance with the national guidelines for the ethical conduct of research involving human subjects (2008).
Clinical Trial Registration
This research does not involve any clinical trials
Permission to reproduce material from other sources
Not Applicable
Authors’ Contribution:
- Safia Khalil Ali: Methodology, Data collection & Analysis, Writing – Original Draft
- Elshazali Widaa Ali: Conceptualization, Supervision, Writing – Review & Editing, Acceptance of final version
References
- Libani IV, Guy EC, Melchiori L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112(3):875–885.
CrossRef - Hagag AA, Elfrargy MS, Gazar RA, El-Lateef AE. Therapeutic value of combined therapy with deferasirox and silymarin on iron overload in children with beta-thalassemia. Mediterr J Hematol Infect Dis. 2013;5:e2013065.
CrossRef - Hershko C. Pathogenesis and management of iron toxicity in thalassemia. Ann N Y Acad Sci. 2010;1202:1–9.
CrossRef - Breuer W, Ghoti H, Shattat A, et al. Non-transferrin bound iron in thalassemia: differential detection of redox active forms in children and older patients. Am J Hematol. 2012;87(1):55–61.
CrossRef - Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011;118(13):3479–3488.
CrossRef - Bresgen N, Eckl PM. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules. 2015;5(2):808–847.
CrossRef - Karunaratna A, Ranasingha JS, Mudiyanse RM. Iron overload in beta-thalassemia major patients. Int J Blood Transfus Immunohematol. 2017;7:33–40.
CrossRef - Taher AT, Saliba AN. Iron overload in thalassemia: different organs at different rates. Hematology Am Soc Hematol Educ Program. 2017;2017(1):265–271.
CrossRef - Arab-Zozani M, Kheyrandish S, Rastgar A, Miri-Moghaddam E. A systematic review and meta-analysis of stature growth complications in β-thalassemia major patients. Ann Glob Health. 2021;87(1):16.
CrossRef - Shah FT, Sayani F, Trompeter S, DrasarE, Piga Challenges of blood transfusions in β-thalassemia. Blood Rev. 2019;37:100588.
CrossRef - Pinto VM, Forni GL. Management of iron overload in beta-thalassemia patients: clinical practice update based on case series. Int J Mol Sci. 2020;21(24):8771.
CrossRef - Ebeid FS, Khan NI. The adverse impact of thalassemia major on adolescents’ oral health-related quality of life. J Pediatr Hematol Oncol. 2020;42(5):e345–e351.
CrossRef - Soltanpour MS, Hagh MF, Kamali K, Jafari GA, Davari K. Frequency of C282Y and H63D mutations of HFE gene and their correlation with iron status in Iranian beta-thalassemia major patients. Iran J Pediatr Hematol Oncol. 2017;7(4):154–162.
- Derchi G, Dessì C, Bina P, et al. Risk factors for heart disease in transfusion-dependent thalassemia: serum ferritin revisited. Intern Emerg Med. 2019;14(3):365–370.
CrossRef - Martins R, Silva B, Proença D, Faustino P. Differential HFE gene expression is regulated by alternative splicing in human tissues. PLoS One. 2011;6(3):e17542.
CrossRef - Hollerer I, Bachmann A, Muckenthaler MU. Pathophysiological consequences and benefits of HFE mutations: 20 years of research. Haematologica. 2017;102(5):809–817.
CrossRef - Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(1):328–343.
CrossRef - Asif S, Begemann M, Raza S. Polycythemia in patients with hereditary hemochromatosis: real or myth? J Clin Med Res. 2019;11(6):422–427.
CrossRef - Radwan A, Othman I. Hereditary hemochromatosis and JAK2-positive polycythemia vera. Clin Case Rep. 2021;9:e04907.
CrossRef - Oliveira SC, Sousa MD, Pinto JP. ER stress and iron homeostasis: a new frontier for the UPR. Biochem Res Int. 2011;2011:896474.
CrossRef - Silva B, Martins R, Proença D, Fleming R, Faustino P. The functional significance of E277K and V295A HFE mutations. Br J Haematol. 2012;158(3):399–408.
CrossRef - Gerhard GS, Chokshi R, Still CD, et al. The influence of iron status and genetic polymorphisms in the HFE gene on the risk for postoperative complications after bariatric surgery: a prospective cohort study in 1,064 patients. Patient Saf Surg. 2011;5(1):1.
CrossRef - Aranda N, Viteri FE, Montserrat C, Arija V. Effects of C282Y, H63D, and S65C HFE gene mutations, diet, and lifestyle factors on iron status in a general Mediterranean population from Tarragona, Spain. Ann Hematol. 2010;89(8):767–773.
CrossRef - Rahmani R, Naseri P, Safaroghli-Azar A, Tarighi S, Hosseini T, Hojjati MT. Investigation of correlation between H63D and C282Y mutations in HFE gene and serum ferritin level in beta-thalassemia major patients. Transfus Clin Biol. 2019;26(4):249–252.
CrossRef - Camaschella C, Pagani A. Advances in understanding iron metabolism and its crosstalk with erythropoiesis. Br J Haematol. 2018;182(4):481–494.
CrossRef - Mishra AK, Tiwari A. Iron overload in beta thalassaemia major and intermedia patients. Maedica. 2013;8(4):328–332.
- Veríssimo MP, Loggetto SR, Fabron Junior A, et al. Brazilian Thalassemia Association protocol for iron chelation therapy in patients under regular transfusion. Rev Bras Hematol Hemoter. 2013;35(6):428–434.
CrossRef - Economou M, Printza N, Teli A, et al. Renal dysfunction in patients with beta-thalassemia major receiving iron chelation therapy either with deferoxamine and deferiprone or with deferasirox. Acta Haematol. 2010;123(3):148–152.
CrossRef - Al-Kuraishy HM, Al-Gareeb AI. Comparison of deferasirox and deferoxamine effects on iron overload and immunological changes in patients with blood transfusion-dependent β-thalassemia. Asian J Transfus Sci. 2017;11(1):13–17.
CrossRef - Jaouni SKA. Survival and disease complications of thalassemia major: experience of 14 years at King Abdulaziz University Hospital, Jeddah, KSA. JKAU: Med Sci. 2010;17:19–28.
CrossRef - Shaw J, Chakraborty A, Nag A, Chattopadyay A, Dasgupta AK, Bhattacharyya M. Intracellular iron overload leading to DNA damage of lymphocytes and immune dysfunction in thalassemia major patients. Eur J Haematol. 2017;99(5):399–408.
CrossRef - Diyah Permatasari T, Dwi Kartikasari G, Ismail C. Correlation between adherence therapy of iron chelation levels with serum ferritin levels in major beta-thalassemia patients at Kediri District General Hospital. Indones Health J. 2023;2(1):38–43.
CrossRef - Aguilar-Martinez P, Bismuth M, Blanc F, et al. The southern French registry of genetic hemochromatosis: a tool for determining clinical prevalence of the disorder and genotype penetrance. Haematologica. 2010;95(4):551–556.
CrossRef - Madani HA, Afify RA, Abd El-Aal AA, Salama N, Ramy N. Role of HFE gene mutations on developing iron overload in beta-thalassemia carriers in Egypt. East Mediterr Health J. 2011;17(6):546–551.
CrossRef - AlFadhli S, Salem M, Shome DK, Mahdi N, Nizam R. The effects of HFE polymorphisms on biochemical parameters of iron status in Arab beta-thalassemia patients. Indian J Hematol Blood Transfus. 2017;33(4):545–551.
CrossRef - Bradbury R, Fagan E, Payne SJ. Two novel polymorphisms (E277K and V212V) in the haemochromatosis gene HFE. Hum Mutat. 2000;15(1):120.
CrossRef - Karimi M, Yavarian M, Delbini P, et al. Spectrum and haplotypes of the HFE hemochromatosis gene in Iran: H63D in beta-thalassemia major and the first E277K homozygous. Hematol J. 2004;5(6):524–527.
CrossRef - Mendes AI, Ferro A, Martins R, et al. Non-classical hereditary hemochromatosis in Portugal: novel mutations identified in iron metabolism-related genes. Ann Hematol. 2009;88(3):229–234.
CrossRef - Campos WN, Massaro JD, Martinelli AD, Mendes-Junior CT, Donadi EA. Genetic structure of coding region of the HFE gene: SNPs, haplotypes and suggested allele nomenclature. Hum Immunol. 2013;74(1):143.
CrossRef - Mikhailova SV, Babenko VN, Ivanoshchuk DE, et al. Haplotype analysis of the HFE gene among populations of Northern Eurasia, in patients with metabolic disorders or stomach cancer, and in long-lived people. BMC Genet. 2016;17(1):83.
CrossRef
Abbreviations lIst
βTM- Beta-thalassemia major; bp- Base pairs; CBC- Complete blood count; DNA- Deoxyribonucleic acid; Hb- Hemoglobin; HH- Hereditary hemochromatosis; HFE- Iron regulatory gene/protein; ICT- Iron chelation therapy; IOC(s)- Iron overload complication(s); NTBI- Non-transferrin-bound iron; PCR- Polymerase chain reaction; SD- Standard deviation; SF- Serum ferritin; SNV– Single nucleotide variant; SPSS- Statistical package for social sciences.

