
Manuscript accepted on :23-06-2025
Published online on: 30-06-2025
Plagiarism Check: Yes
Reviewed by: Dr. Huzef U and Dr. Kulvinder Kaur
Second Review by: Dr. Marwah Mohmmed
Final Approval by: Dr. Patorn Piromchai
Muhammad Najmi Munawwar Mohd Hatta1 and Normala Abd Latip1,2*
and Normala Abd Latip1,2*
1Department of Pharmacology and Pharmaceutical Life sciences, Faculty of Pharmacy, Universiti Teknologi MARA (UiTM) Cawangan Selangor, Puncak Alam, Malaysia.
2Department Integrative Pharmacogenomics Institute (iPROMISE), Universiti Teknologi MARA (UiTM) Cawangan Selangor, Puncak Alam, Malaysia.
Corresponding Author E-mail:drnormala6351@uitm.edu.my
DOI : https://dx.doi.org/10.13005/bpj/3162
Abstract
Cytochrome P450 2B6 (CYP2B6) plays a significant role in the metabolism of various drugs, yet its regulation and clinical relevance are often underappreciated. Numerous substrates of CYP2B6 have been identified to date, and these findings may contribute to potential drug-drug interactions in patients. Extensive research has been conducted to assess how drugs affect the activity of CYP2B6 and, consequently, impact therapy effectiveness in patients. These investigations are crucial for establishing safe drug dosages for patients. The objective of this review is to offer insights into the drug-drug interactions associated with CYP2B6 and to provide an overview of its impact on drug administration in patients. This review systematically explores CYP2B6 transcriptional control, polymorphism-dependent activity, and drug-drug interaction (DDI) potential. We emphasize the influence of nuclear receptors, including CAR, PXR, HNF3β, and oestrogen receptor on CYP2B6 gene expression, as well as the consequences of allelic variants such as *6, *16, and *18 on drug metabolism. Using a PRISMA-guided literature search, we synthesized 96 original studies addressing modulators of CYP2B6 and their clinical implications. Our findings demonstrate that regulatory mechanisms and genetic diversity significantly shape interindividual differences in CYP2B6-mediated drug metabolism. Clinically important substrates like efavirenz, cyclophosphamide, artemisinin, and tamoxifen exhibit interaction profiles shaped by both enzyme induction and inhibition. This review evaluates CYP2B6-mediated interactions through the lens of nuclear receptor-driven transcriptional control and polymorphism-associated enzyme variability.
Keywords
Constitutive androstane receptor (CAR); CYP2B6; Inducer; Inhibitor; Pregnane X receptor (PXR)
Download this article as:| Copy the following to cite this article: Hatta M. N. M. M, Latip N. A. Comprehensive Review of CYP2B6 Transcriptional Modulators, Variants, and Their Role in Drug Interactions. Biomed Pharmacol J 2025;18(2). |
| Copy the following to cite this URL: Hatta M. N. M. M, Latip N. A. Comprehensive Review of CYP2B6 Transcriptional Modulators, Variants, and Their Role in Drug Interactions. Biomed Pharmacol J 2025;18(2). Available from: https://bit.ly/44rasH0 |
Introduction
Cytochrome P450 enzymes are essential in xenobiotic metabolism, with CYP2B6 contributing to the clearance of ~8% of clinically used drugs despite its low hepatic abundance. The enzyme is responsible for metabolizing a structurally diverse range of compounds including antiretrovirals, antimalarials, antidepressants, and anticancer agents. However, CYP2B6 activity exhibits high interindividual variability, largely attributed to both genetic polymorphisms and regulation by nuclear receptors.1
Antiretroviral, antimalarial, antineoplastic, anticonvulsant and steroids are some of the groups that have been identified metabolised by CYP2B6. When treating specific diseases, simultaneous administration of drugs with varying actions may be utilized to achieve synergistic effects. However, some of these drugs may induce or inhibit the concentration of cytochrome P450 enzymes such as CYP2B6, thereby impacting their ability to metabolize the drugs. Consequently, it is imperative to thoroughly evaluate this issue to ensure the safe administration of doses and drug combinations to patients.2
While previous reviews, such as Hedrich et al., have described CYP2B6 substrates and drug-drug interactions, mechanistic insights into its transcriptional regulation and the combined effect of nuclear receptor activation and genetic variants remain underexplored.
CYP2B6 is characterized by high genetic variability, impacting transcriptional regulation, catalytic activity, splicing, and protein expression. there are 38 CYP2B6 allele variants, each with distinct functional effects ranging from no function to normal, decreased, or increased CYP2B6 activity. Notably, CYP2B6*1, considered the wild type allele, demonstrates normal CYP2B6 function, a characteristic shared with *2, *5, and *17 alleles.3,4
The review goes beyond conventional focus on CAR and PXR by incorporating emerging regulators like HNF3β and oestrogen receptors, thus offering a deeper understanding of the transcriptional complexity of CYP2B6. Additionally, it uniquely emphasizes the clinical relevance of polymorphic variants such as *6, *16, and *18, linking them to specific drug interactions and therapeutic outcomes. This integrative approach, combining transcriptional regulation, pharmacogenetics, and drug interaction data, provides a valuable resource for advancing personalized pharmacotherapy.
This review aims to (1) delineate CYP2B6 regulation via nuclear receptors, (2) evaluate the clinical significance of its polymorphisms, and (3) systematically catalogue CYP2B6 inducers and inhibitors relevant to clinical DDI risk. By doing so, we provide an updated, mechanistic perspective on the implications of CYP2B6 modulation in personalized pharmacotherapy. We aim for this review to serve as a valuable resource for further research and aid in determining appropriate drug dosages metabolized by CYP2B6 in the treatment of various diseases.
Materials and Methods
A systematic literature search was conducted using two electronic databases: Scopus and Web of Science (WoS). The search strategy included the following keywords: “Cytochrome P450 2B6” AND “drug interaction”. The search was completed without date restrictions and included studies available in English, yielding 109 records after initial retrieval. No duplicate records or ineligible entries were identified by automation tools or manual screening at the initial stage.
All 109 records were screened for relevance based on titles and abstracts. No records were excluded at this stage. Subsequently, all records were sought for full-text retrieval, and none were reported as not retrievable.
The 109 full-text articles were assessed for eligibility based on predefined inclusion and exclusion criteria. Articles were excluded if they were review papers (n = 12) or non-research notes (n = 1). Following the eligibility assessment, a total of 96 original research articles were deemed suitable and included in the final review.
This selection process followed the PRISMA 2020 guidelines and is illustrated in the PRISMA flow diagram (Figure 1).
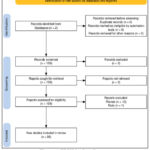 |
Figure 1: PRISMA flow diagramClick here to view Figure |
Results
Overview of Included Studies
Using PRISMA-compliant methodology, 96 original articles were included. These studies encompassed in vitro enzyme assays, human hepatocyte models, clinical pharmacokinetic data, and genotyping analyses. The majority explored CYP2B6 modulation by therapeutic agents, while a subset investigated genetic variation and its metabolic implications.
Transcriptional Regulation of CYP2B6
The transcriptional regulation of CYP2B6 exhibits inter- and intra-individual variability across the human population.5 Numerous transcription factors, including the constitutive androstane receptor (CAR), 6,7 pregnane X receptor (PXR), 4,8 and other nuclear receptors, 7,9–12 have been identified as controlling CYP2B6 expression.
However, these receptors alone do not account for the complete regulation, as studies show their overexpression does not induce CYP2B6 in non-hepatic cells. Additional nuclear receptors such as hepatocyte nuclear factor 3β (HNF3β), glucocorticoid receptor, and oestrogen receptor have been identified to contribute to CYP2B6 gene expression.
Polymorphisms and Allelic Variants
Thirty-eight CYP2B6 allelic variants have been identified to date, with *6, 16, and 18 being the most clinically relevant. CYP2B66 is associated with reduced clearance of efavirenz and elevated plasma concentrations.13–16 Conversely, its role in cyclophosphamide bioactivation is enhanced, potentially increasing therapeutic toxicity.17 CYP2B6*18, prevalent in African populations, encodes a nonfunctional enzyme, while *16 also exhibits reduced activity. Co-occurrence of *16 and *18 significantly elevates the risk of adverse drug reactions due to impaired metabolism.3,18,19
Clinical Significance of CYP2B6 Polymorphisms
CYP2B6 polymorphisms significantly affect enzyme activity and drug metabolism. The CYP2B66 variant, found in 15–60% of populations, is linked to reduced efavirenz metabolism, increasing the risk of adverse CNS effects and drug resistance.15 Conversely, CYP2B6*6 may enhance the bioactivation of cyclophosphamide. Other clinically relevant variants include CYP2B6*18, which lacks enzymatic activity and is common among African populations, and CYP2B6*16, also associated with reduced function. Co-occurrence of these alleles can lead to elevated drug levels and toxicity.
Substrate Spectrum of CYP2B6
CYP2B6 metabolizes structurally diverse substrates, including:
Antiretrovirals (efavirenz, nevirapine)
Antineoplastics (cyclophosphamide, tamoxifen)
Antimalarials (artemisinin)
Antidepressants (bupropion, selegiline)
Anesthetics (propofol)
Table 1: CYP2B6 clinically important substrates
| Pharmacological class | Drugs | Major metabolic pathways | References |
| Benzodiazepines | DiazepamClotiazepam | DemethylationHydroxylation | 20,21 |
| MAO inhibitors | Selegiline | Demethylation | 22,23 |
| Antidepressant | Bupropion | Hydroxylation | 24,25 |
| Antiretroviral | NevirapineEfavirenz | HydroxylationHydroxylation | 2616,27–29 |
| Anticancer | TamoxifenCyclophosphamide
Ifosfamide |
HydroxylationHydroxylation
Hydroxylation |
30,3132,33
34 |
| Antimalarial | Artemisinin | Hydroxylation | 35,36 |
| Opioids | PethidineMethadone | DemethylationDemethylation | 3738 |
| Anesthetics | PropofolRopivacaine | HydroxylationHydroxylation | 3940 |
| Antiepilepsy | Mephobarbital | Demethylation | 41 |
| Steroids | Testosterone | Hydroxylation | 42 |
Inducers and Inhibitors of CYP2B6
Several drugs can modulate CYP2B6 activity either through induction or inhibition. Enzyme induction generally results in increased drug metabolism, reducing plasma levels and potentially decreasing efficacy. Conversely, inhibition may increase plasma concentrations, enhancing toxicity risks. Drugs such as efavirenz and cyclophosphamide are both substrates and inducers of CYP2B6. Thiotepa and tamoxifen act as irreversible inhibitors, while sertraline, selegiline, and ticlopidine function as potent reversible or mechanism-based inhibitors.
Table 2: CYP2B6 inducers and inhibitors
| Inducer | Inhibitor |
| Efavirenz | Thiotepa |
| Nevirapine | Tamoxifen |
| Cyclophosphamide | Crizotinib |
| Artemisinin | Tamoxifen |
| Carbamazepine | Selegiline |
| Phenytoin | Sertraline |
| Phenobarbital | Clopidogrel |
| Clotrimazole | Ticlopidine |
| Rifampin | Itraconazole |
| Troglitazone | Voriconazole |
| Dexamethasone | Orphenadrine |
| Estradiol | Desmethylazelastine |
| Amlodipine | |
| Barnidipine | |
| Colchicine | |
| Amiodarone | |
| Furafylline | |
| Ethynylestradiol |
Drug-Induced Modulation
Numerous drugs serve as inducers (e.g., phenobarbital, rifampin, artemisinin) or inhibitors (e.g., thiotepa, tamoxifen, ticlopidine) of CYP2B6. Induction often occurs via PXR or CAR activation, leading to enhanced clearance of co-administered CYP2B6 substrates. In contrast, mechanism-based inhibition, such as that exerted by thiotepa or sertraline, may reduce metabolic capacity and raise toxicity risks.43–45
Discussion
CYP2B6 transcriptional regulation
The regulatory mechanisms of CYP2B6 expression are governed by an intricate network of nuclear receptors, transcriptional co-factors, and hepatocyte-enriched elements. Among these, the constitutive androstane receptor (CAR) and pregnane X receptor (PXR) are primary mediators, responding to a wide array of xenobiotics.7,9,46 However, co-regulation with HNF3β, glucocorticoid receptor, and oestrogen receptor contributes to the dynamic and tissue-specific regulation of CYP2B6 expression.7,11,12 Activation of these receptors by certain drugs can lead to altered CYP2B6 expression, thereby contributing to variability in drug response and potential drug-drug interactions. Understanding these regulatory mechanisms is essential for predicting such interactions. This complexity may explain variability in CYP2B6 induction across individuals and contexts.
Although the principal transcription factors implicated in the regulation of CYP2B6 are the CAR and PXR receptors. It’s noted that these receptors alone cannot fully account for CYP2B6 transcription regulation. While they moderately induce target genes such as CYP2Cs, overexpression of these receptors fails to induce CYP2B6 expression in non-hepatic cell lines.47 Recent studies have indicated the involvement of other nuclear receptors in CYP2B6 expression, contributing to inter- and intra-individual variation. Nuclear receptors like hepatocyte nuclear factor 3β can endogenously induce CYP2B6 expression, further adding to this variation.
Constitutive androstane receptor (CAR) and Pregnane X receptor (PXR)
Constitutive androstane receptor (CAR) and Pregnane X receptor (PXR) are predominantly expressed in the liver and intestine, with minimal expression in other tissues. These nuclear receptors play crucial roles in both oxidative phase I and conjugative phase II metabolism, as well as in the transport of drugs out of cells and as efflux pumps for drugs that are substrates to multidrug resistance (MDR1) protein.
Compared to steroid hormone receptors, both CAR and PXR exhibit low affinity and binding specificity. This characteristic makes them well-suited for recognizing various chemicals continuously entering the body. CAR, as implied by its name, remains constitutively active without the need for ligand binding. 48 In contrast, PXR requires activation by various compounds such as glucocorticoids, antibiotics, and antifungals, as it is a ligand-dependent specificity receptor.9
CAR is primarily located in the cytoplasm, forming a protein complex with heat shock protein (hsp90). Its activation occurs upon translocation into the nucleus. Following translocation, CAR heterodimerizes with retinoid x receptor α (RXRα) and binds to response elements, thereby initiating the transcription of target genes such as CYP enzymes. Meanwhile, PXR undergoes activation similar to CAR but necessitates ligands for activation, as mentioned earlier.
Hepatocyte Nuclear Factor 3β
The regulation of CYP2B6 expression extends beyond the well-characterized nuclear receptors such as CAR and PXR, incorporating the influence of hepatocyte-enriched transcription factors like hepatocyte nuclear factor 3β (HNF3β), also known as FOXA2.49 Recent studies have demonstrated that HNF3β plays a pivotal role in modulating CYP2B6 transcription in human liver cells.
HNF3β involved in liver development and function, particularly in regulating hepatic genes such as CYP2B6. This transcription factor directly modulates the expression and activity of CYP2B6. HNF3β binds directly to specific regions within the CYP2B6 promoter, facilitating chromatin remodeling and enabling the recruitment of other nuclear receptors, thus enhancing transcriptional activity.50
In the context of liver-specific gene regulation, HNF3β acts as a pioneer factor that can modulate the accessibility of DNA to other transcriptional regulators. Li et al. showed through chromatin immunoprecipitation and promoter-luciferase assays that overexpression of HNF3β significantly increases CYP2B6 promoter activity, while knockdown of HNF3β leads to a marked reduction in its expression. This indicates that HNF3β is both necessary and sufficient for optimal CYP2B6 transcription, particularly in hepatocytes.49,51,52
Furthermore, HNF3β appears to work synergistically with CAR and PXR. While CAR and PXR require activation by xenobiotic ligands to upregulate CYP2B6, HNF3β provides the foundational chromatin accessibility that allows these ligand-activated receptors to bind effectively.52 This cooperative interaction between HNF3β and xenobiotic nuclear receptors may explain the tissue-specific and interindividual variability observed in CYP2B6 expression levels.
In summary, HNF3β is a critical upstream regulator of CYP2B6, operating both independently and in cooperation with classical nuclear receptors. Its presence ensures hepatic specificity and basal promoter activation, laying the groundwork for further induction by xenobiotics. These findings underscore the importance of considering transcriptional co-regulators like HNF3β in understanding drug metabolism variability and in developing more precise pharmacogenetic models.
Glucocorticoid Receptors (GR)
The GR, a ligand-activated transcription factor, is known to interact with other nuclear receptors such as constitutive androstane receptor (CAR) and pregnane X receptor (PXR), forming a regulatory network that modulates the expression of various cytochrome P450 genes, including CYP2B6.
Although GR does not directly bind to the CYP2B6 promoter as its primary activator, its role is permissive and synergistic, it enhances the transcriptional activity of CAR and PXR. This is achieved through mechanisms such as coactivator recruitment, chromatin remodeling, or post-translational modifications that increase the nuclear translocation and DNA binding efficiency of CAR and PXR. In particular, co-treatment with glucocorticoids like dexamethasone has been shown to amplify CAR-mediated induction of CYP2B6, suggesting a facilitative role of GR in the CAR-CYP2B6 axis.53,54
Recent findings have further clarified the role of GR in the indirect enhancement of CYP2B6 expression.53–57 A 2023 study by Pascussi et al. highlighted that GR activation via dexamethasone significantly boosts CYP2B6 expression in primary human hepatocytes only when CAR is co-expressed,57 reinforcing the notion that GR functions as a modulator rather than a primary driver. Moreover, GR signaling contributes to CYP2B6 induction during inflammatory and stress responses, which is relevant in clinical contexts involving corticosteroid therapy.
Taken together, the glucocorticoid receptor plays an essential auxiliary role in CYP2B6 regulation, primarily by facilitating the action of other nuclear receptors such as CAR and PXR. This interaction is clinically relevant, as co-administration of glucocorticoids may alter the metabolism of drugs that are CYP2B6 substrates, necessitating careful consideration in polypharmacy and personalized medicine.
Oestrogen Receptors (ER)
Cytochrome P450 2B6 (CYP2B6) expression is influenced by a variety of other nuclear receptors, including the oestrogen receptors (ERs), particularly ERα. These receptors, upon binding to oestrogens such as 17β-estradiol, can modulate the transcription of CYP2B6 either directly via estrogen response elements (EREs) or indirectly through cross-talk with other nuclear receptors like CAR (constitutive androstane receptor) and PXR (pregnane X receptor).58,59
Evidence suggests that ERα enhances CYP2B6 expression synergistically with CAR, especially in hepatocytes. A pivotal study by Koh et al. (2012) demonstrated that high concentrations of estradiol can significantly upregulate CYP2B6 mRNA and protein levels in human liver cells. This induction was observed to be CAR-dependent, as co-transfection of ERα and CAR resulted in markedly increased transcriptional activity at the CYP2B6 promoter region.60 The study identified a composite ERα/CAR response element within the promoter, supporting a cooperative regulatory mechanism.
Moreover, oestrogen regulation of CYP2B6 has clinical implications, particularly in pregnancy, hormonal therapy, and sex-based pharmacokinetics. For instance, during pregnancy, elevated oestrogen levels have been associated with increased CYP2B6 activity, potentially affecting the metabolism of drugs like efavirenz and bupropion, which are CYP2B6 substrates.59
Recent insights from bioinformatics and transcriptomic analyses have expanded this understanding. A 2023 systems biology study by Zhang et al. integrated human liver RNA-seq data and identified sex hormone-related nuclear receptor pathways, including ER signaling, as key modulators of inter-individual variability in CYP2B6 expression, particularly among female donors.61,62
In summary, oestrogen receptors, especially ERα, indirectly but significantly contribute to the regulation of CYP2B6 by enhancing the transcriptional activity of CAR and possibly PXR. This regulation may underlie sex-specific differences in drug metabolism, and understanding these mechanisms is critical for optimizing therapies in hormone-sensitive populations.
Other Nuclear Receptors
Another important co-regulator is Retinoid X receptor alpha (RXR-alpha), also known as NR2B1 is a nuclear receptor that in humans is encoded by the RXRA gene. RXRα forms heterodimers with CAR and PXR, which is essential for DNA binding and activation of CYP2B6 transcription. RXRα enhances ligand-dependent and constitutive activation of CAR/PXR target genes, including CYP2B6. Recent structural studies have shown that RXRα’s interaction with various nuclear receptors can significantly alter gene activation potential depending on ligand availability and cellular context.63
Emerging evidence also points to peroxisome proliferator-activated receptors (PPARs) and liver X receptor (LXR) as modulators of CYP2B6. PPARα activation was shown to indirectly modulate CYP2B6 expression via metabolic cross-talk in hepatocytes, particularly under fasting or lipid-loading conditions.64,65 Similarly, LXRα, a sensor of cholesterol and oxysterols, has been implicated in hepatic detoxification pathways, and recent data suggest it may regulate a subset of CYP genes, including CYP2B6, especially in response to lipid-rich diets.66,67
CYP2B6 clinical significance polymorphism
CYP2B6 exhibits significant genetic variation, impacting various aspects such as transcription regulation, catalytic activity, splicing, and protein expression. To date, there are 38 distinct variants of the CYP2B6 allele, each with differing functional consequences ranging from complete loss of function to normal function, and even increased or decreased activity levels of the CYP2B6 enzyme. Among these variants, CYP2B6*1 is considered the wild type allele, functioning similarly to *2, *5, and *17 alleles.15,62,68,69
Clinically, polymorphic variants such as *6 and 18 substantially affect enzyme kinetics and drug response. For example, CYP2B66 alters efavirenz clearance, necessitating dose adjustments to avoid neurotoxicity. Conversely, the same variant enhances cyclophosphamide bioactivation, which may increase cytotoxic efficacy but also toxicity risk.17,61,70–72 The presence of null alleles like *18 underscores the need for genotype-guided therapy, especially in populations with high allele frequencies.
CYP2B6*6 Variant
CYP2B6 plays a pivotal role in the clearance of efavirenz from the body. Responsible for the hydroxylation of efavirenz to its inactive metabolite, 8-hydroxyefavirenz. Efavirenz constitutes a key component of highly active antiretroviral therapy (HAART) and is recommended as initial therapy in HAART regimens.13,14
CYP2B6*6 has emerged as the most clinically significant variant affecting the metabolism of the anti-HIV drug efavirenz. This variant allele is observed at frequencies ranging from 15% to 60% across different human populations. The CYP2B6*6 variant exhibits an increased Km compared to the wild-type CYP2B6*1, resulting in reduced efavirenz metabolism.62 In HIV patients harbouring the CYP2B6*6 variant, elevated plasma concentrations of efavirenz can occur, potentially leading to central nervous system side effects. This, in turn, may contribute to patient non-adherence and increase the risk of developing drug resistance.
Bioactivation of cyclophosphamide has been linked to the CYP2B6 enzyme which converts it to 4-hydroxycyclophosphamide. CYP2B6*6 has a significant impact on the hydroxylation of cyclophosphamide with an increase of metabolic activity. 73 Like cyclophosphamide, ifosfamide also activated by CYP3A4 and CYP2B6 equally to its active metabolite which is 4-hydroxyifosfamide. CYP2B6*6 has been linked to ifosfamide-induced encephalopathy due to reduced activity of the enzyme that shifts the pathway of ifosfamide to form chloroacetaldehyde. 74
Other variants
CYP2B6*18 has been reported to be an allele without enzymatic activity75. It is most common in African population, and it has no noticeable enzymatic activity when tested with bupropion, artemether and selegiline. 3,76 This is believed to happen due to change of hydrophobic isoleucine to polar uncharged threonine at position 328 of amino acid that alters CYP2B6 hydrophobicity.
Another variant that is dominant among the African population is CYP2B6*16 which results in reduced activity of the enzyme. If a patient bears both variants which are CYP2B6*18 and *16, this will lead to a significant increase of efavirenz plasma concentration up to fivefold than the normal patient. 1 As a result, patients will have a high risk in developing adverse effects.
CYP2B6 substrates
CYP2B6 was thought to have minimal involvement in xenobiotic metabolism and was mistakenly believed to play a minor role in drug metabolism rather than having significant substrates of its own. However, advancements in the discovery and development of specific antibodies over the past decade have facilitated the identification of more substrates for CYP2B6.
Presently, CYP2B6 is recognized as a crucial enzyme in drug metabolism, including drugs such as bupropion, artemisinin, propofol, and efavirenz, among others as listed in Table 1. Despite the increasing ease of identifying CYP2B6 substrates, the precise extent of xenobiotics metabolized by CYP2B6 remains uncertain. CYP2B6 is responsible for approximately 8% of drug metabolism in the current market.
CYP2B6 inducer, inhibitor and drug-drug interaction
Drugs can interact with cytochrome enzymes in various ways; some interactions involve only one isozyme, while others may affect multiple isozymes, as seen with methadone, which is metabolized by both CYP3A4 and CYP2B6.38,77,78 With the discovery of more substrates for CYP2B6, there is an increased likelihood of drug-drug interactions, especially when this isozyme shares substrates with other enzymes such as CYP3A4, CYP2C9, and CYP2C19.2,79,80
Drug-drug interactions related to cytochrome enzymes occur when one of the drugs taken by a patient induces or inhibits enzyme activity. In general, enzyme induction leads to increased drug metabolism, resulting in reduced plasma levels and potentially rendering the drug ineffective. However, in the case of prodrugs, enzyme induction can lead to an accumulation of metabolites to potentially harmful levels. Conversely, enzyme inhibition reduces drug metabolism, leading to higher drug plasma levels, which can result in overdose, toxicity, and, in extreme cases, death, depending on the drugs involved.
It is important to note that some substrates metabolized by CYP2B6 may also induce or inhibit its activity. For example, cyclophosphamide and selegiline, which are metabolized by CYP2B6, also act as a CYP2B6 inducer and inhibitor, respectively. A comprehensive list of CYP2B6 inducers and inhibitors is provided in Table 2.
Antiretroviral
Efavirenz, a prominent antiretroviral, is primarily metabolized by CYP2B6, and its interaction with CYP2B6 polymorphism is well-established. In addition to efavirenz, nevirapine is also notable for being predominantly metabolized by CYP2B6. Both drugs have the ability to induce their own metabolism, a phenomenon known as autoinduction, resulting in increased clearance. Consequently, they can reduce the plasma concentration of other antiretrovirals, and substrates metabolized by CYP2B6.
The autoinduction of CYP2B6 by efavirenz and nevirapine occurs through weak activation of the pregnane X receptor (PXR) and strong activation of the constitutive androstane receptor (CAR) receptor. With repeated doses of efavirenz, a mixed induction and inhibition effect may occur, ultimately resulting in a net induction of CYP2B6 activity.
Ritonavir, despite not being a substrate for CYP2B6, functions as an inhibitor of this enzyme. However, adjusting the dose of bupropion is necessary when ritonavir and lopinavir are taken concurrently, as exposure to bupropion and its active metabolite, hydroxybupropion, decreases. This induction is attributed to the activation of the pregnane X receptor (PXR) and the increased expression of UGT1A1 protein, which enhances the glucuronidation process.38,81
Antineoplastic
Cyclophosphamide, widely used in cancer treatment, is an antineoplastic agent metabolized by CYP2B6. It is a prodrug requiring activation by the CYP system to produce phosphoramide mustard and acrolein, highly cytotoxic agents that disrupt DNA synthesis in cancer cells. Notably, cyclophosphamide can induce its own metabolism by activating the PXR pathway.
Another substrate of CYP2B6 is thiotepa, an alkylating agent primarily used in the treatment of breast, bladder, and ovarian cancer. While thiotepa is only minimally metabolized by CYP2B6, it undergoes significant metabolism by CYP3A4 into its active metabolite, TEPA.43 Thiotepa is also recognized as a potent inhibitor of CYP2B6, inhibiting cyclophosphamide metabolism to 4-hydroxycyclophosphamide by approximately 80% . Unlike reversible inhibitors, the potency of thiotepa’s inhibition not only depends on dosage but also on the frequency and duration of administration, posing a significant risk.
Another irreversible inhibitor is tamoxifen, widely used in the treatment of breast cancer. Its metabolite, 4-hydroxytamoxifen, has been identified as responsible for the inactivation of CYP2B6 when incubated with NADPH and tamoxifen. Attempts to restore the activity of the inactivated CYPs by adding fresh reductase have failed, leading to the conclusion that tamoxifen acts as an irreversible inhibitor of CYP2B6. 82,83
Crizotinib, an anticancer medication utilized in the treatment of non-small cell lung carcinoma, undergoes metabolism primarily by CYP3A4. It has been noted that crizotinib exhibits moderate inhibition of CYP2B6. 84 Consequently, caution is advised when administering drugs metabolized by CYP2B6 to prevent potential drug-drug interactions with crizotinib.
Cisplatin, employed in the treatment of various cancers such as lung, breast, ovarian, and testicular cancer, has been reported to inhibit CYP2B6 activity.85 However, the degree of inhibition is relatively low and clinically insignificant compared to other drugs. It inhibits approximately 25% of CYP2B6 activity, categorizing it as a weak inhibitor of CYP2B6.
Antimalarial
In the context of multidrug resistance, artemisinin has emerged as the primary agent for treating malaria. While it is predominantly metabolized by CYP2B6, the precise mechanism remains unclear. Two artemisinin compounds, Qinghaosu (artemisinin) and dihydroartemisinin, neither inhibits nor induces CYP2B6, whereas artemisinin has the potential to induce CYP2B6.86
The induction of CYP2B6 and CYP3A4 enzyme activity were observed after administration of oral artemisinin-piperaquine fixed combination dose.87 The autoinduction of artemisinin suggests a heightened potential for drug-drug interactions if other drugs metabolized by the same isozyme are administered concurrently.
Anticonvulsant
Anticonvulsant is one of the prominent classes that showed drug-drug interaction not only in CYP2B6 but in other CYP450 isozymes. Carbamazepine and phenytoin are moderate inducers for CYP2B6 while phenobarbital is a strong inducer for CYP2B6 which all are also CYP3A4 inducer.88 Most of the CYP3A4 inducers has been reported to be inducer for CYP2B6.53,89
Phenobarbital, an antiepileptic medication, is well-known for its association with the induction of numerous CYP isozymes. The mRNA, protein, and activity of CYP2B6 is significantly induced by phenobarbital in human liver slices. 90 Phenobarbital has been tested on the metabolism of cyclophosphamide and ifosfamide and the result was positive as it increased the conversion of both drugs into their active metabolites 91. This activation is due to the activation of the PXR receptor predominantly. 68
Carbamazepine functions as a CYP2B6 inducer through the activation of the constitutive androstane receptor (CAR). This activation is weakly associated with the pregnane X receptor (PXR), suggesting CAR as a potential regulator of CYP2B6 induction.
Phenytoin induces the metabolism of ifosfamide via CYP2B6. It has been demonstrated to act as an inducer for CYP2B6 rather than CYP3A4. Consequently, pretreatment with phenytoin may enhance the efficacy of ifosfamide therapy. Similar to carbamazepine, phenytoin is believed to induce its effects through CAR activation rather than PXR.
Antidepressant
Selegiline is an antidepressant from the monoamine oxidase inhibitor group, also commonly used as antiparkinson drug, recognized to be majorly metabolized by CYP2B6. 92,93 Selegiline could inhibit the activity of CYP2B6 through inhibition of bupropion into hydroxybupropion when taken concomitantly.94
Sertraline, a selective serotonin reuptake inhibitor (SSRI), is another antidepressant that is metabolized by CYP2B6.95,96 Sertraline is a strong CYP2B6 inhibitor 45 and will inhibit the metabolism of efavirenz up to 48%. Despite these reports, no significant drug-drug interactions have been demonstrated or confirmed in clinical settings, which leaves the matter unclear.
Anticoagulant
Apart from coumarin, there are no other report on anticoagulant metabolized by CYP2B6.1 However, anticoagulants clopidogrel and ticlop, are strongly associated with CYP2B6.97,98 Both drugs are prodrugs and depend on CYPs isozymes to be effective. An in vitro study showed that clopidogrel and ticlopidine are potent and irreversible inhibitor for CYP2B6 through mechanism-based inhibition.99 The chemically reactive thiolactone metabolites of clopidogrel and ticlopidine are responsible for the CYP2B6 inhibition.100 Reduction of ketamine metabolism has been shown when taken concurrently with ticlopidine.101 Thus, ticlopidine and clopidogrel are regarded as a strong CYP2B6 inhibitors and should be considered in terms of drug-drug interaction with other CYP2B6 substrates.102
Antifungal
No antifungal has been reported as a substrate of CYP2B6. However, some antifungals are purported to induce CYP2B6 activity. Through in vitro studies, clotrimazole has been identified as a potent CYP2B6 inducer. It is thought to activate the human pregnane X receptor (PXR), thereby upregulating the expression of CYP2B6.68
Itraconazole is considered as an inhibitor for CYP2B6 and may potentially produce drug-drug interaction.103 But the metabolism of tramadol has been reported to be insignificantly inhibited by itraconazole. This is probably due to the fact that tramadol also metabolised by CYP2D6 and only a small portion by CYP2B6.104 Voriconazole has been reported to be responsible for competitive inhibition for four CYPs isozymes including CYP2B6 through an increase of efavirenz exposure and potential side effects.105
Several antifungal agents have been shown to modulate the activity and expression of cytochrome P450 2B6 (CYP2B6), leading to clinically relevant drug–drug interactions. These interactions can occur through inhibition, induction, or mechanism-based inactivation of CYP2B6, affecting the metabolism of co-administered drugs such as bupropion, efavirenz, cyclophosphamide, and methadone.
One of the most studied azole antifungals in this context is ketoconazole, a potent inhibitor of multiple CYP isoforms. Ketoconazole has been shown to inhibit CYP2B6 activity non-specifically, although it is not as potent against CYP2B6 as it is against CYP3A4.24,106 However, at higher concentrations, ketoconazole can significantly suppress CYP2B6-mediated metabolism, which may lead to increased plasma levels of CYP2B6 substrates and potential toxicity.
Voriconazole, a second-generation triazole antifungal, is a substrate and inhibitor of CYP2C19, CYP2C9, and CYP3A4, but also inhibits CYP2B6 to a lesser extent. Studies have shown that voriconazole can modestly inhibit CYP2B6 activity in vitro, and co-administration with CYP2B6 substrates may necessitate dosage adjustments, especially for drugs with narrow therapeutic indices.64,107,108
Another antifungal agent, clotrimazole, has been shown to act as a mechanism-based inhibitor (MBI) of CYP2B6. MBI occurs when a compound is metabolized into a reactive intermediate that irreversibly inactivates the enzyme. Clotrimazole binds and inactivates CYP2B6 in a time- and concentration-dependent manner, which can profoundly suppress enzyme activity even after the drug is cleared from circulation.82
While less is known about the effects of fluconazole on CYP2B6, evidence suggests it is a weak inhibitor of this isoform.109 Clinical relevance may be limited unless combined with other inhibitors or in genetically poor metabolizers of CYP2B6 substrates.51,65
In summary, azole antifungals such as ketoconazole, voriconazole, and clotrimazole can inhibit CYP2B6 activity, potentially leading to altered drug metabolism and drug-drug interactions. Awareness of these interactions is essential, particularly in patients receiving multiple medications or those treated with narrow therapeutic window drugs metabolized by CYP2B6.
Other Classes of Drugs
Rifampin is an antituberculosis and not a substrate for CYP2B6. However, rifampin induces CYP2B6 activity by activating both the pregnane X receptor (PXR) and constitutive androstane receptor (CAR) receptors.89 This induction has been observed to enhance the plasma clearance of methadone and diminish the efficacy of efavirenz.110, 111 Therefore, dose adjustments should be carefully considered when administering both drugs concurrently with rifampin.
Two antihistamines, orphenadrine and desmethylazelastine, have been identified as inhibitors of CYP2B6. Orphenadrine has been shown to inhibit the metabolism of artemisinin by up to 76%.87 Similarly, desmethylazelastine, a metabolite of azelastine, has been found to non-competitively inhibit CYP2B6 activity.112
One oral antidiabetic medication within the thiazolidinedione group, troglitazone, has been reported to induce CYP2B6 activity. A significant rise in CYP2B6 immunoreactive protein levels was observed 113. However, this finding was not substantiated by Yamazaki et al.,114 who discovered that troglitazone actually inhibited the catalytic activity of CYP2B6 rather than inducing it.
Amlodipine and barnidipine have been demonstrated as potent inhibitors of CYP2B6 through the inhibition of the 7-benzyloxyresorufin O-dealkylation process. However, this evidence is limited to an in vitro study, and the inhibition of CYP2B6 by calcium channel blockers has only been predicted in in vivo animal model.115
Colchicine is a drug used to treat gout and it has been shown to suppress the activity of CYP2B6. It is reported by Dvorak et al to alter PXR, CAR and RXR signal transduction and lead to CYP2B6 inhibition.116 Additional inducers of CYP2B6 include dexamethasone and the human hormone oestradiol, as reported in various studies.58,117. Conversely, inhibitors for CYP2B6 include amiodarone, furafylline, and ethynylestradiol. However, these drugs lack substantial clinical evidence, and the potential for causing drug-drug interactions remains uncertain.
Conclusion
In summary, the regulation of CYP2B6 activity represents a complex interplay between transcriptional mechanisms and genetic polymorphisms, each contributing uniquely to inter-individual variability in drug metabolism. This dual-layered control system not only dictates baseline enzyme expression but also dynamically responds to xenobiotic exposure through a broad spectrum of nuclear-receptor and transcription-factor pathways, including pregnane X receptor (PXR), constitutive androstane receptor (CAR), estrogen receptor (ER), glucocorticoid receptor (GR), hepatocyte nuclear factor-3β (HNF-3β), and retinoid X receptor (RXR). Consequently, CYP2B6 plays a central role in mediating clinically significant drug-drug interactions (DDIs), particularly in polypharmacy settings where enzyme inducers, inhibitors, and auto-inducers coexist.
Importantly, the co-occurrence of pharmacogenetic variants such as CYP2B6*6, *16, and *18 with potent enzyme modulators (e.g., efavirenz, thiotepa) can amplify or mitigate metabolic effects, thereby altering therapeutic efficacy or toxicity profiles. These insights emphasize the necessity for genotype-guided therapeutic strategies, especially in vulnerable populations receiving CYP2B6-metabolized drugs.
The paradoxical behavior of some drugs that function as both substrates and inducers further complicates treatment regiments and highlights the limitations of one-size-fits-all dosing. Clinical implementation of pharmacogenomic screening, combined with an understanding of nuclear receptor biology, could transform how clinicians anticipate metabolic liability and tailor interventions.
As the field advances, integrating CYP2B6 regulatory insights into therapeutic drug monitoring, drug development, and clinical decision support tools will be essential. Future studies should aim to close the translational gap between in vitro findings and real-world pharmacokinetic outcomes, enabling safer and more effective drug therapy across diverse patient populations.
Acknowledgment
The author would like to thank Universiti Teknologi MARA (UiTM) for granting this research work.
Funding Sources
This research was supported/partially supported by Faculty of Pharmacy and RMC UiTM research grant FRGS/1/2022/SKK15/UITM/02/6 and 600-UITMSEL (PI. 5/4)
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinicial Trial Registration
This research does not involve any clinical trials.
Permission to Reproduce Material from Other Sources
Not Applicable
Authors Contributions
- Muhammad Najmi Munawwar: Conceptualization, Methodology, Writing – Original Draft.
- Normala Abd Latip: Data Collection, Analysis, Writing – Review & Editing.
References
- Hedrich WD, Hassan HE, Wang H. Insights into CYP2B6-mediated drug-drug interactions. Acta Pharm Sin B. 2016;6(5):413-425.
CrossRef - Mutalib NA, Rafi MAAM, Latip NA. Revisiting CYP2C9-Mediated drug-drug Interactions: A Review. Res J Pharm Technol. 2021;14(11):6166-6172.
CrossRef - Klein K, Lang T, Saussele T, et al. Genetic variability of CYP2B6 in populations of African and Asian origin: Allele frequencies, novel functional variants, and possible implications for anti-HIV therapy with efavirenz. Pharmacogenet Genomics. 2005;15(12):861-873.
CrossRef - Lamba V, Lamba J, Yasuda K, et al. Hepatic CYP2B6 Expression: Gender and Ethnic Differences and Relationship to CYP2B6 Genotype and CAR (Constitutive Androstane Receptor) Expression. Journal of Pharmacology and Experimental Therapeutics. 2003;307(3):906-922.
CrossRef - Hembrom PS, Jose J, Shalini K, Grace T. Detection of variation in expression of insecticide resistance gene Cyp4d2 involved in detoxification of insecticides in drosophila melanogaster. Res J Pharm Technol. 2019;12(11):5315-5319.
CrossRef - Lang T, Klein K, Fischer J, et al. Extensive genetic polymorphism in the human CYP2B6 gene with impact on expression and function in human liver. Pharmacogenetics. 2001;11(5):399-415.
CrossRef - Choi HS, Chung M, Tzameli I, et al. Differential transactivation by two isoforms of the orphan nuclear hormone receptor CAR. Journal of Biological Chemistry. 1997;272(38):23565-23571.
CrossRef - Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA. Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol. 2001;60(3):427-431.
CrossRef - Honkakoski P, Sueyoshi T, Negishi M. Drug-activated nuclear receptors CAR and PXR. Ann Med. 2003;35(3):172-182.
CrossRef - Wang H, Negishi M. Transcriptional Regulation of Cytochrome P450 2B Genes by Nuclear Receptors. Curr Drug Metab. 2005;4(6):515-525.
CrossRef - Li L, Li D, Heyward S, Wang H. Transcriptional regulation of CYP2B6 expression by hepatocyte nuclear factor 3β in human liver cells. PLoS One. 2016;11(3):1-15.
CrossRef - Dumas B, Harding HP, Choi HS, et al. A new orphan member of the nuclear hormone receptor superfamily closely related to Rev-Erb. Molecular Endocrinology. 1994;8(8):996-1005.
CrossRef - Tsuchiya K, Gatanaga H, Tachikawa N, et al. Homozygous CYP2B 6*6 (Q172H and K262R) correlates with high plasma efavirenz concentrations in HIV-1 patients treated with standard efavirenz-containing regimens. Biochem Biophys Res Commun. 2004;319(4):1322-1326.
CrossRef - Martín AS, Gómez AI, García-Berrocal B, et al. Dose reduction of efavirenz: An observational study describing cost-effectiveness, pharmacokinetics and pharmacogenetics. Pharmacogenomics. 2014;15(7):997-1006.
CrossRef - Nolan D, Phillips E, Mallal S. Efavirenz and CYP2B6 Polymorphism: Implications for Drug Toxicity and Resistance. Clinical Infectious Diseases. 2006;42(3):408-410.
CrossRef - Destra Z, Saussele T, Ward B, et al. Impact of CYP2B6 polymorphism on hepatic efavirenz metabolism in vitro. Pharmacogenomics. 2007;8(6):547-558.
CrossRef - Joshua Matthew Abbott, Diane Calinski and PH. Metabolism of Cyclophosphamide by the Human Cytochrome P450 2B6 . The Faseb Journal.
- Che Sulaiman IS, Basri M, Chan KW, et al. African Journal of Pharmacy and Pharmacology In vitro antioxidant, cytotoxic and phytochemical studies of Clinacanthus nutans Lindau leaf extracts. Afr J Pharm Pharmacol. 2015;9(34):861-874.
CrossRef - Hussain MS. Patient counseling about herbal-drug interactions, African Journal of Traditional. Complementary and Alternative Medicines. 2011;8(5):152-163.
CrossRef - Yang TJ, Krausz KW, Shou M, et al. Inhibitory monoclonal antibody to human cytochrome P450 2B6. Biochem Pharmacol. 1998;55(10):1633-1640.
CrossRef - Niwa T, Shiraga T, Ishii I, Kagayama A, Takagi A. Contribution of human hepatic cytochrome P450 isoforms to the metabolism of psychotropic drugs. Biol Pharm Bull. 2005;28(9):1711-1716.
CrossRef - Shin HS. Metabolism of selegiline in humans. Identification, excretion, and stereochemistry of urine metabolites. Drug Metab Dispos. 1997;25(6):657-662.
- Taavitsainen P, Anttila M, Nyman L, Karnani H, Salonen JS, Pelkonen O. Selegiline Metabolism and Cytochrome P450 Enzymes : In vitro Study in Human Liver Microsomes *. 2000;1998:215-221.
CrossRef - Turpeinen M, Zanger UM. Cytochrome P450 2B6: Function, genetics, and clinical relevance. Drug Metabol Drug Interact. 2012;27(4):185-197.
CrossRef - Hogeland GW, Swindells S, McNabb JC, Kashuba ADM, Yee GC, Lindley CM. Lopinavir/ritonavir reduces bupropion plasma concentrations in healthy subjects. Clin Pharmacol Ther. 2007;81(1):69-75.
CrossRef - Erickson DA, Mather G, Trager WF, Levy RH, Keirns JJ. Characterization of the in vitro biotransformation of the HIV-1 reverse transcriptase inhibitor nevirapine by human hepatic cytochromes P-450. Drug Metabolism and Disposition. 1999;27(12):1488-1495.
CrossRef - Soeria-Atmadja S, Österberg E, Gustafsson LL, et al. Genetic variants in CYP2B6 and CYP2A6 explain interindividual variation in efavirenz plasma concentrations of HIV-infected children with diverse ethnic origin. PLoS One. 2017;12(9).
CrossRef - M. R, S. C, H. F, et al. Influence of CYP2B6 polymorphism on plasma and intracellular concentrations and toxicity of efavirenz and nevirapine in HIV-infected patients. Pharmacogenet Genomics. 2005;15(1):1-5.
CrossRef - Tsuchiya K, Gatanaga H, Tachikawa N, et al. Homozygous CYP2B 6*6 (Q172H and K262R) correlates with high plasma efavirenz concentrations in HIV-1 patients treated with standard efavirenz-containing regimens. Biochem Biophys Res Commun. 2004;319(4):1322-1326.
CrossRef - Coller JK, Krebsfaenger N, Klein K, et al. The influence of CYP2B6, CYP2C9 and CYP2D6 genotypes on the formation of the potent antioestrogen Z-4-hydroxy-tamoxifen in human liver. Br J Clin Pharmacol. 2002;54(2):157-167.
CrossRef - Styles JA, Davies A, Lim CK, et al. Genotoxicity of tamoxifen epoxide and toremifene in human lymphoblastoid cells containing human cytochrome P450s. Carcinogenesis. 1994;15(1):5-9.
CrossRef - Lindley C, Hamilton G, McCune JS, et al. The effect of cyclophosphamide with and without dexamethasone on cytochrome P450 3A4 and 2B6 in human hepatocytes. Drug Metabolism and Disposition. 2002;30(7):814-822.
CrossRef - Huang Z, Roy P, Waxman DJ. Role of human liver microsomal CYP3A4 and CYP2B6 in catalyzing N- dechloroethylation of cyclophosphamide and ifosfamide. Biochem Pharmacol. 2000;59(8):961-972.
CrossRef - Roy P, Tretyakov O, Wright J, Waxman DJ. Stereoselective metabolism of ifosfamide by human P-450S 3A4 and 2B6. Favorable metabolic properties of R-enantiomer. Drug Metabolism and Disposition. 1999;27(11):1309-1318.
CrossRef - Simonsson USH, Jansson B, Hai TN, Huong DX, Tybring G, Ashton M. Artemisinin autoinduction is caused by involvement of cytochrome P450 2B6 but not 2C9. Clin Pharmacol Ther. 2003;74(1):32-43.
CrossRef - Svensson USH, Ashton M. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br J Clin Pharmacol. 1999;48(4):528-535.
CrossRef - Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of hepatic and intestinal cytochrome P450 3A and 2B6 in the metabolism, disposition, and miotic effects of methadone. Clin Pharmacol Ther. 2004;76(3):250-269.
CrossRef - Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of hepatic and intestinal cytochrome P450 3A and 2B6 in the metabolism, disposition, and miotic effects of methadone. Clin Pharmacol Ther. 2004;76(3):250-269.
CrossRef - Court MH, Duan SX, Hesse LM, Venkatakrishnan K, Greenblatt DJ. Cytochrome P-450 2B6 is responsible for interindividual variability of propofol hydroxylation by human liver microsomes. Anesthesiology. 2001;94(1):110-119.
CrossRef - Al YO et. Metabolism of a New Local Anesthetic, Ropivacaine, by Human Hepatic Cytochrome P450. Anesthesiology. 1995;31(4):305-309.
- Kobayashi K, Abe S, Nakajima M, et al. Role of human CYP2B6 in S-mephobarbital N-demethylation. Drug Metabolism and Disposition. 1999;27(12):1429-1433.
CrossRef - Imaoka S, Yamada T, Hiroi T, et al. Multiple forms of human P450 expressed in Saccharomyces cerevisiae systematic characterization and comparison with those of the rat. Biochem Pharmacol. 1996;51(8):1041-1050.
CrossRef - Jacobson P, Green K, Birnbaum A, Remmel R. Cytochrome P450 isozymes 3A4 and 2B6 are involved in the in vitro human metabolism of thiotepa to TEPA. Cancer Chemother Pharmacol. 2002;49(6):461-467.
CrossRef - Chiba K, Kobayashi K, Ishizuka T, Shimada N, Yoshimura Y, Kamijima K. Sertraline N-demethylation is catalyzed by multiple isoforms of human cytochrome P-450 in vitro. Drug Metabolism and Disposition. 1999;27(7):763-766.
CrossRef - Obach RS, Cox LM, Tremaine LM. Sertraline is metabolized by multiple cytochrome P450 enzymes, monoamine oxidases, and glucuronyl transferases in human: An in vitro study. Drug Metabolism and Disposition. 2005;33(2):262-270.
CrossRef - Pramodh B, Ashok Kumar M, Shanmugasundaram P. Prospective observational study on drug use evaluation of antiplatelet agents in tertiary care hospital. Res J Pharm Technol. 2017;10(12):4328-4332.
CrossRef - Gervot L. Human CYP2B6: expression, inducibility and catalytic activities. Lippincott Williams & Wilkins. 1999;9:295-306.
CrossRef - Rastogi H, Sharma G, Sharma A, Jana S. In-vitro assessment of the inhibitory effects of dietary flavones on the regulated metabolism of CYP 450: Potential for herb-drug interactions. Res J Pharm Technol. 2020;13(12):6086-6092.
CrossRef - Li L, Li D, Heyward S, Wang H. Transcriptional regulation of CYP2B6 expression by hepatocyte nuclear factor 3β in human liver cells. PLoS One. 2016;11(3). doi:10.1371/journal.pone.0150587
CrossRef - Faucette SR, Sueyoshi T, Smith CM, Negishi M, Lecluyse EL, Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. Journal of Pharmacology and Experimental Therapeutics. 2006;317(3):1200-1209.
CrossRef - Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: Crosstalk and consequences. Annu Rev Pharmacol Toxicol. 2008;48:1-32. doi:10.1146/annurev.pharmtox.47.120505.105349
CrossRef - Dickmann LJ, Isoherranen N. Quantitative prediction of CYP2B6 induction by estradiol during pregnancy: Potential explanation for increased methadone clearance during pregnancy. Drug Metabolism and Disposition. 2013;41(2):270-274.
CrossRef - Wang H, Faucette SR, Gilbert D, et al. Glucocorticoid receptor enhancement of pregnane X receptor-mediated CYP2B6 regulation in primary human hepatocytes. Drug Metabolism and Disposition. 2003;31(5):620-630.
CrossRef - Sugatani J, Nishitani S, Yamakawa K, et al. Transcriptional regulation of human UGT1A1 gene expression: Activated glucocorticoid receptor enhances constitutive androstane receptor/ pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol Pharmacol. 2005;67(3):845-855.
CrossRef - Li Y, Buckley D, Wang S, Klaassen CD, Zhong XB. Genetic polymorphisms in the TATA box and upstream phenobarbital-responsive enhancer module of the UGT1A1 promoter have combined effects on UDP-glucuronosyltransferase 1A1 transcription mediated by constitutive androstane receptor, pregnane X receptor, or glucocorticoid receptor in human liver. Drug Metabolism and Disposition. 2009;37(9):1978-1986.
CrossRef - Dvořák Z, Modrianský M, Pichard-Garcia L, et al. Colchicine down-regulates cytochrome P450 2B6, 2C8, 2C9, and 3A4 in human hepatocytes by affecting their glucocorticoid receptor-mediated regulation. Mol Pharmacol. 2003;64(1):160-169.
CrossRef - Y MM, Maurel P, Pascussi J marc. Colchicine Down-Regulates Cytochrome P450 2B6, 2C8, 2C9, and 3A4 in Human Hepatocytes by Affecting Their Glucocorticoid Receptor-Mediated Regulation. Mol Pharmacol. 2003;64(1):160-169.
CrossRef - Koh KH, Jurkovic S, Yang K, et al. Estradiol induces cytochrome P450 2B6 expression at high concentrations: Implication in estrogen-mediated gene regulation in pregnancy. Biochem Pharmacol. 2012;84(1):93-103.
CrossRef - Dickmann LJ, Isoherranen N. Quantitative prediction of CYP2B6 induction by estradiol during pregnancy: Potential explanation for increased methadone clearance during pregnancy. Drug Metabolism and Disposition. 2013;41(2):270-274.
CrossRef - Koh KH, Jurkovic S, Yang K, et al. Estradiol induces cytochrome P450 2B6 expression at high concentrations: Implication in estrogen-mediated gene regulation in pregnancy. Biochem Pharmacol. 2012;84(1):93-103.
CrossRef - Calinski D, Zhang H, Sridar C, Hollenberg PF. Metabolism of cyclophosphamide by the human cytochrome P450 2B6 polymorphic variants. FASEB Journal.
- Zhang H, Sridar C, Kenaan C, Amunugama H, Ballou DP, Hollenberg PF. Polymorphic variants of cytochrome P450 2B6 (CYP2B6.4-CYP2B6.9) exhibit altered rates of metabolism for bupropion and efavirenz: A charge-reversal mutation in the K139E variant (CYP2B6.8) impairs formation of a functional cytochrome P450-reductase complex. Journal of Pharmacology and Experimental Therapeutics. 2011;338(3):803-809.
CrossRef - Evans RM, Mangelsdorf DJ. Nuclear receptors, RXR, and the big bang. Cell. 2014;157(1):255-266.
CrossRef - Guan S, Huang M, Li X, Chen X, Chan E, Zhou SF. Intra- and inter-ethnic differences in the allele frequencies of cytochrome P450 2B6 gene in Chinese. Pharm Res. 2006;23(9):1983-1990.
CrossRef - Zhou SF, Xue CC, Yu XQ, Li C, Wang G. Clinically Important Drug Interactions Potentially Involving Mechanism-Based Inhibition of Cytochrome P450 3A4 and the Role of Therapeutic Drug Monitoring. Pharm Res. 2006;23(9):1983-1990.
CrossRef - Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. 2009;2(5-6):231-237.
CrossRef - Huang Z, Roy P, Waxman DJ. Role of human liver microsomal CYP3A4 and CYP2B6 in catalyzing N- dechloroethylation of cyclophosphamide and ifosfamide. Biochem Pharmacol. 2000;59(8):961-972.
CrossRef - Faucette SR, Wang H, Hamilton GA, et al. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metabolism and Disposition. 2004;32(3):348-358.
CrossRef - Nolan D, Phillips E, Mallal S. Efavirenz and CYP2B6 polymorphism: Implications for drug toxicity and resistance. Clinical Infectious Diseases. 2006;42(3):408-410.
CrossRef - Xie HJ, Yasar Ü, Lundgren S, et al. Role of polymorphic human CYP2B6 in cyclophophamide bioactivation. Pharmacogenomics Journal. 2003;3(1):53-61.
CrossRef - Rae JM, Soukhova N V, Flockhart DA. P450 2B6 : Implications for Cyclophosphamide Metabolism. 2002;30(5):525-530.
CrossRef - Mutalib NA, Latip NA. Antagonistic Drug-herb Interactions between Clinacanthus nutans and Cyclophosphamide on WRL 68 Cell Line. Pharmaceutical Sciences and Research. 2020;7(2):81-89.
CrossRef - Xie HJ, Yasar Ü, Lundgren S, et al. Role of polymorphic human CYP2B6 in cyclophophamide bioactivation. Pharmacogenomics Journal. 2003;3(1):53-61.
CrossRef - Palareti G, Legnani C, Cosmi B, et al. Comparison between different D-Dimer cutoff values to assess the individual risk of recurrent venous thromboembolism: Analysis of results obtained in the DULCIS study. Int J Lab Hematol. 2016;38(1):42-49.
CrossRef - Klein K, Lang T, Saussele T, et al. Genetic variability of CYP2B6 in populations of African and Asian origin: Allele frequencies, novel functional variants, and possible implications for anti-HIV therapy with efavirenz. Pharmacogenet Genomics. 2005;15(12):861-873.
CrossRef - Zanger UM, Klein K. Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): Advances on polymorphisms, mechanisms, and clinical relevance. Front Genet. 2013;4(MAR):1-12.
CrossRef - Totah RA, Sheffels P, Roberts T, Whittington D, Thummel K, Kharasch ED. Role of CYP2B6 in stereoselective human methadone metabolism. Anesthesiology. 2008;108(3):363-374.
CrossRef - Richards-Waugh LL, Primerano DA, Dementieva Y, Kraner JC, Rankin GO. Fatal methadone toxicity: Potential role of CYP3A4 genetic polymorphism. J Anal Toxicol. 2014;38(8):541-547.
CrossRef - Hummel MA, Locuson CW, Gannett PM, et al. CYP2C9 genotype-dependent effects on in vitro drug-drug interactions: Switching of benzbromarone effect from inhibition to activation in the CYP2C9.3 variant. Mol Pharmacol. 2005;68(3):644-651.
CrossRef - Mahdi F, Raza ST, Rizvi S, Abbas S, Karoli R. Distribution of Genetic Polymorphisms in Drug Metabolizing Gene Cytochrome P450 (CYP2C8*3 and CYP2C9*2) in a North Indian Type 2 Diabetes Population. Explor Res Hypothesis Med. 2016;1(3):42-46.
CrossRef - Kharasch ED, Mitchell D, Coles R, Blanco R. Rapid clinical induction of hepatic cytochrome P4502B6 activity by ritonavir. Antimicrob Agents Chemother. 2008;52(5):1663-1669.
CrossRef - Sridar C, Kent UTEM, Notley LM, Gillam EMJ, Hollenberg PF. Effect of Tamoxifen on the Enzymatic Activity of Human Cytochrome CYP2B6. 2002;301(3):945-952.
CrossRef - Coller JK, Krebsfaenger N, Klein K, et al. The influence of CYP2B6, CYP2C9 and CYP2D6 genotypes on the formation of the potent antioestrogen Z-4-hydroxy-tamoxifen in human liver. Br J Clin Pharmacol. 2002;54(2):157-167.
CrossRef - Gallucci G, Tartarone A, Lombardi L, Aieta M. When crizotinib-induced bradycardia becomes symptomatic: Role of concomitant drugs. Expert Rev Anticancer Ther. 2015;15(7):761-763.
CrossRef - Masek V, Anzenbacherová E, Machová M, Brabec V, Anzenbacher P. Interaction of antitumor platinum complexes with human liver microsomal cytochromes P450. Anticancer Drugs. 2009;20(5):305-311.
CrossRef - Xing J, Kirby BJ, Whittington D, Wan Y, Goodlett DR. Evaluation of P450 inhibition and induction by artemisinin antimalarials in human liver microsomes and primary human hepatocytes. Drug Metabolism and Disposition. 2012;40(9):1757-1764.
CrossRef - Svensson USH, Ashton M. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br J Clin Pharmacol. 1999;48(4):528-535.
CrossRef - Faucette SR, Wang H, Hamilton GA, et al. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metabolism and Disposition. 2004;32(3):348-358.
CrossRef - Faucette SR, Sueyoshi T, Smith CM, Negishi M, Lecluyse EL, Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. Journal of Pharmacology and Experimental Therapeutics. 2006;317(3):1200-1209.
CrossRef - Sarsat J pierre, Waziers I De, Housset C, Balladur P, Beaune P, Lerche-langrand C. Induction of Cytochrome P450 2B6 and 3A4 Expression by Phenobarbital and Cyclophosphamide in Cultured Human Liver Slices. Pharm Res. 2003;20(4):557-568.
CrossRef - Chang TKH, Yu L, Maurel P, Waxman DJ. Enhanced cyclophosphamide and ifosfamide activation in primary human hepatocyte cultures: Response to cytochrome P-450 inducers and autoinduction by oxazaphosphorines. Cancer Res. 1997;57(10):1946-1954.
- Benetton SA, Fang C, Yang Y ou, et al. P450 phenotyping of the metabolism of selegiline to desmethylselegiline and methamphetamine. Drug Metab Pharmacokinet. 2007;22(2):78-87.
CrossRef - Shin HS. Metabolism of selegiline in humans: Identification, excretion, and stereochemistry of urine metabolites. Drug Metabolism and Disposition. 1997;25(6):657-662.
- Sridar C, Kenaan C, Hollenberg PF. Inhibition of bupropion metabolism by selegiline: Mechanism-based inactivation of human CYP2B6 and characterization of glutathione and peptide adducts. Drug Metabolism and Disposition. 2012;40(12):2256-2266.
CrossRef - Chiba K, Kobayashi K, Ishizuka T, Shimada N, Yoshimura Y, Kamijima K. Sertraline N-demethylation is catalyzed by multiple isoforms of human cytochrome P-450 in vitro. Drug Metabolism and Disposition. 1999;27(7):763-766.
CrossRef - Obach RS, Cox LM, Tremaine LM. Sertraline is metabolized by multiple cytochrome P450 enzymes, monoamine oxidases, and glucuronyl transferases in human: An in vitro study. Drug Metabolism and Disposition. 2005;33(2):262-270.
CrossRef - Chen Y, Liu HF, Liu L, Nguyen K, Jones EB, Fretland AJ. The in vitro metabolism of bupropion revisited: Concentration dependent involvement of cytochrome P450 2C19. Xenobiotica. 2010;40(8):536-546.
CrossRef - Nishiya Y, Hagihara K, Ito T, et al. Mechanism-Based Inhibition of Human Cytochrome P450 2B6 by Ticlopidine , Clopidogrel , and the Thiolactone Metabolite of Prasugrel. 2009;37(3):589-593.
CrossRef - Richter T, Mu TE, Heinkele G, et al. Potent Mechanism-Based Inhibition of Human CYP2B6 by Clopidogrel and Ticlopidine. 2004;308(1):189-197.
CrossRef - Nishiya Y, Hagihara K, Ito T, et al. Mechanism-Based Inhibition of Human Cytochrome P450 2B6 by Ticlopidine , Clopidogrel , and the Thiolactone Metabolite of Prasugrel. 2009;37(3):589-593.
CrossRef - Peltoniemi MA, Saari TI, Hagelberg NM, et al. Exposure to Oral S-ketamine is unaffected by itraconazole but greatly increased by ticlopidine. Clin Pharmacol Ther. 2011;90(2):296-302.
CrossRef - Tornio A, Filppula AM, Niemi M, Backman JT. Clinical Studies on Drug–Drug Interactions Involving Metabolism and Transport: Methodology, Pitfalls, and Interpretation. Clin Pharmacol Ther. 2019;105(6):1345-1361.
CrossRef - Walsky RL, Astuccio A V., Obach RS. Evaluation of 227 drugs for in vitro inhibition of cytochrome P450 2B6. J Clin Pharmacol. 2006;46(12):1426-1438.
CrossRef - Saarikoski T, Saari TI, Hagelberg NM, et al. Effects of terbinafine and itraconazole on the pharmacokinetics of orally administered tramadol. Eur J Clin Pharmacol. 2015;71(3):321-327.
CrossRef - Jeong S, Nguyen PD, Desta Z. Comprehensive in vitro analysis of voriconazole inhibition of eight cytochrome P450 (CYP) enzymes: Major effect on CYPs 2B6, 2C9, 2C19, and 3A. Antimicrob Agents Chemother. 2009;53(2):541-551.
CrossRef - Turpeinen M, Jouko U, Jorma J, Olavi P. Multiple P450 substrates in a single run: Rapid and comprehensive in vitro interaction assay. European Journal of Pharmaceutical Sciences. 2005;24(1):123-132.
CrossRef - Zhou W, Chen MH, Shi W. Influence of phthalates on glucose homeostasis and atherosclerosis in hyperlipidemic mice. BMC Endocr Disord. 2015;15:13.
CrossRef - Wu X, Zhou D, Song J, Sun M, Pan Y. Molecular probes for identification of UDP-glucuronosyltransferase 1 gene polymorphisms for gilbert’s syndrome diagnosis. Biomedical Research (India). 2018;29(10):2111-2115.
CrossRef - Li L, Li D, Heyward S, Wang H. Transcriptional regulation of CYP2B6 expression by hepatocyte nuclear factor 3β in human liver cells. PLoS One. 2016;11(3).
CrossRef - McIlleron HM, Schomaker M, Ren Y, et al. Effects of rifampin-based antituberculosis therapy on plasma efavirenz concentrations in children vary by CYP2B6 genotype. Aids. 2013;27(12):1933-1940.
CrossRef - Totah RA, Sheffels P, Roberts T, Whittington D, Thummel K, Kharasch ED. Role of CYP2B6 in stereoselective human methadone metabolism. Anesthesiology. 2008;108(3):363-374.
CrossRef - Nakajima M, Ohyama K, Nakamura S, Shimada N, Yamazaki H, Yokoi T. Inhibitory effects of azelastine and its metabolites on drug oxidation catalyzed by human cytochrome P-450 enzymes. Drug Metabolism and Disposition. 1999;27(7):792-797.
CrossRef - Sahi J, Hamilton GŒ, Sinz M, et al. Effect of troglitazone on cytochrome P450 enzymes in primary cultures of human and rat hepatocytes. Published online 2000:273-284.
CrossRef - Yamazaki H, Suzuki M, Tane K, Shimada N, Nakajima M, Yokoi T. In vitro inhibitory effects of troglitazone and its metabolites on drug oxidation activities of human cytochrome P450 enzymes: Comparison with pioglitazone and rosiglitazone. Xenobiotica. 2000;30(1):61-70.
CrossRef - Sharma D, Mehta DK, Bhatti K, Das R, Chidurala RM. Amlodipine and atenolol: Combination therapy versus monotherapy in reducing blood pressure – A focus on safety and efficacy. Res J Pharm Technol. 2020;13(6):3007-3013.
CrossRef - Y MM, Maurel P, Pascussi J marc. Colchicine Down-Regulates Cytochrome P450 2B6, 2C8, 2C9, and 3A4 in Human Hepatocytes by Affecting Their Glucocorticoid Receptor-Mediated Regulation. Mol Pharmacol. 2003;64(1):160-169.
CrossRef - Dickmann LJ, Isoherranen N. Quantitative prediction of CYP2B6 induction by estradiol during pregnancy: Potential explanation for increased methadone clearance during pregnancy. Drug Metabolism and Disposition. 2013;41(2):270-274.
CrossRef

