Cisplatin Resistance in Ovarian Cancer: Classical Outlook and Newer Perspectives
 , Prasan Bhandari1, Shraddha Yadav1 and Ashwini Prabhu2
, Prasan Bhandari1, Shraddha Yadav1 and Ashwini Prabhu2 1Department of Pharmacology, Symbiosis Medical College for Women, Symbiosis International University, Pune, India, 412115.
2Yenepoya Research Centre, Yenepoya (Deemed to be University), Mangalore, India, 575018.
Corresponding Author E-mail: asstprof.pharmacology2@smcw.siu.edu.in
DOI : http://dx.doi.org/10.13005/bpj/2297
Download this article as:
![]()
Ovarian cancer is one of the most common gynecological cancers. Recently, there is increase in incidence of ovarian cancer not only India but also worldwide. Ovarian cancer patients exhibit nonspecific symptoms during early course of disease. As a consequence, 70% of these patients are diagnosed in advanced stages. Ovarian cancer treatment includes cytoreductive surgery followed by platinum-based chemotherapy. However, these patients develop fatal recurrence due to development of platinum resistance. Cisplatin, (platinum analog) resistance is multifactorial and complex. Earlier, resistance was mainly attributed to conventional molecular mechanisms like decreased intracellular accumulation of cisplatin, enhanced DNA repair and increased cisplatin detoxification. Nevertheless, emergence of knowledge of tumor biology have lead to discovery of other contributing mechanisms. These tumor microenvironment related factors include physical blockade, hypoxia, cancer stem cells, cancer associated fibroblasts and many others. Understanding these mechanisms of cisplatin resistance is crucial for development of novel strategy to combat the same. Hence, this review summarizes all the mechanisms of resistance of cisplatin in ovarian cancer.
KEYWORDS:Cisplatin Resistance; Molecular Mechanism; Ovarian Cancer; Tumor
Introduction
Ovarian cancer is the third most common cancer among women in India.1 In India approximately, the incidence of ovarian carcinoma is expected to range from 1.7 to 15.2 for 2012 to 2014 in different population-based cancer registers. 1 Moreover, in India, there was a rough estimate of 59,276, as new ovarian cancer cases in 2020.2 According to cancer stat facts, in United states of America, estimated new cases and estimated deaths from ovarian cancer are 21,750 and 13,940 respectively.
Ovarian cancer patients usually have nonspecific symptoms like gaseous distention of stomach, abdominal swelling, changes in bowel habits and unexplained weight loss. 3 Hence, ovarian cancer is often not diagnosed till advanced stages. 3 Moreover, at advanced stages, five year survival rate is as low as 30%. 4 Advanced stage ovarian cancer is treated by cytoreductive surgery followed by chemotherapy with six cycles of platinum containing drug either cisplatin or carboplatin in combination paclitaxel. 3 Majority (i.e70%) of ovarian cancer patients respond to a platinum and taxane based chemotherapy.5 However, 50% of these patients will develop recurrence. 8 This recurrence is due to development and persistence of drug-resistant cells in peritoneal cavity, which eventually grow progressively leading to death of patient. 5,6
Development of resistance to cisplatin can be acquired or intrinsic. 7 The conventionally described mechanisms of drug resistance are decreased uptake, enhanced efflux, increased DNA repair, augmented detoxification by GSH conjugation. 7,8 Relatively novel discoveries include cisplatin resistance due to autophagy induction, tumor microenvironment related factors like hypoxia, cancer associated fibroblasts, macrophages, activation of the EMT pathway. 7,9–11
Understanding all the mechanisms of resistance is crucial in order to develop novel strategies to combat cisplatin resistance. Moreover, understanding of cisplatin resistance mechanisms is important to identify and detect biomarkers. Biomarkers are macromolecules that can be measured and indicate pharmacological response in the body. In this paper, we will review all mechanisms of resistance including those due to tumor microenvironment.
Epidemiology of ovarian cancer
Ovarian carcinoma is fifth major cause of malignancy related mortality in females. It usually affects women over 65 years of age. In 2018, 295,414 cases were identified across the world. 12 However, ovarian cancer is more common in developed nations than developing world. 13 According to cancer stat facts, in United states of America, estimated new cases and estimated deaths from ovarian cancer are 21,750 and 13,940 respectively. In India approximately, the incidence of ovarian carcinoma is expected to range from 1.7 to 15.2 for 2012 to 2014 in different population-based cancer registers. 1 Moreover, in India, there was a rough estimate of 59,276, as new ovarian cancer cases in 2020.2
Pathogenesis of ovarian cancer
“Ovarian cancer” is multiple group of malignancies affecting ovary. Cancer of ovary may arise from one of three types of cell, those are surface epithelial cells, sex cord stromal cells, germ cells. (Fig.1) However, tumors with more than one type of cell are also encountered and these are designated as “mixed” tumors. 14 90% of malignant ovarian cancers are surface epithelial-tumors. Also, referred as epithelial ovarian cancer or carcinoma. Furthermore, WHO classifies surface epithelial-tumors into serous, mucinous and endometroid depending upon cell type. Serous, mucinous, endometroid resemble fallopian tube endometrium, endometrial glands and endocervical epithelium respectively. 15 Also, depending on atypia, these are further classified into benign, malignant, borderline and depending upon tumor subtype into low-grade (type I) and high-grade (type II). 15 High grade serous ovarian carcinoma (HGSOC) accounts for 70% of cases. 16
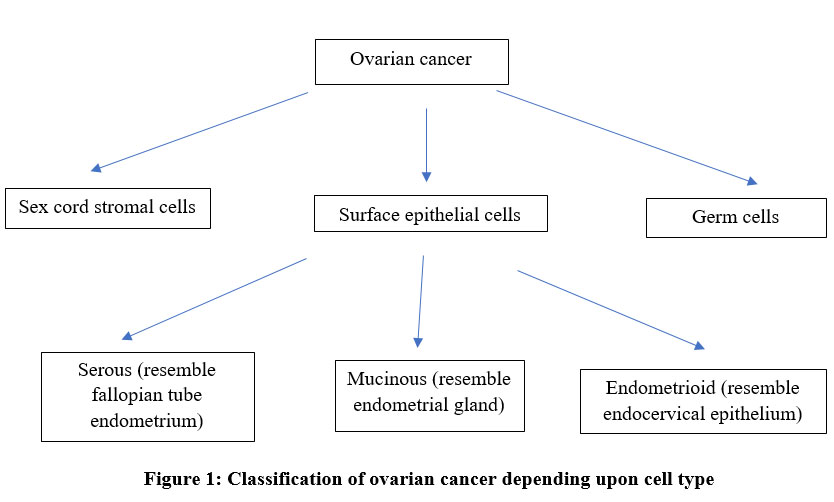 |
Figure 1: Classification of ovarian cancer depending upon cell type. |
The exact etiology of ovarian cancer is not known yet.4 However, two hypothesis for ovarian cancer pathogenesis are proposed – incessant ovulation and fallopian tube theory. In 1971, Fathala, observed that human female have incessant ovulation from puberty till menopause. It was further proposed that during ovulation, trauma occurs to surface epithelium and also damage to DNA, which is ultimately responsible for neoplastic transformation. 13,17 However, this theory fails to explain pathogenesis in patients with polycystic ovarian disease in which there is no incessant ovulation.
Later, cloning of BRCA 1&2 genes led to women undergoing prophylactic bilateral salpingo-oophorectomies to reduce risk of ovarian cancer. Furthermore, study of histology of fallopian tube of these women, revealed surface epithelium of tubes may play an important role in pathogenesis of ovarian cancer. 13,16. Recent studies have revealed, that 60-70% of high grade serous ovarian carcinoma (HGSOC) arise from lesions in fallopian tube. 16
Molecular Features of HGSOC
Approximately 20 % of high grade serous ovarian cancer have BRCA1 & BRCA2 gene mutations. BRCA1/BRCA2 proteins are tumor suppressor proteins involved in DNA damage repair process through homologous recombination (process is explained in detail later in text). One more mutation common in HGSOC is T53, almost 97% of cases. This gene is responsible for DNA repair (homologous recombination), the cell cycle and apoptosis upon irreparable DNA damage. 18 In 50% of HGSOC, T53 and /or BRCA1/2 mutations along with other mutations can result in total deficiency of homologous recombination. 18
Pharmacotherapy of ovarian cancer
Current treatment strategy of ovarian cancer involves surgical debulking, chemotherapy and radiation treatment. Chemotherapy is most critical of all. Chemotherapeutic agents include taxane group (paclitaxel or docetaxel) and platinum containing compounds (cisplatin or carboplatin). Chemotherapy is administered through intravenous route (IV), intraperitoneal route (IP) or both (IV/IP). Although intraperitoneal route offers range of advantages, it is not widely accepted. 19,20
At advanced stage, after surgical debulking, current standard regimen is 6 cycle of carboplatin (AUC 5) and paclitaxel (175 mg/m2) IV over three hours, repeat after every 3 weeks. 3 This regimen of carboplatin + paclitaxel is found to be equally effective as cisplatin + paclitaxel in terms of overall survival & progression free-survival. Moreover, regimen containing carboplatin was found to be more tolerable , as patients experienced less frequently non hematologic side effects like nausea vomiting and neuropathy. 21 However, carboplatin and cisplatin acts by same mechanism, have similar pharmacokinetic properties hence their mechanism of resistance remains same. Furthermore, cisplatin was used widely before carboplatin, hence research till date, is more focused on describing mechanisms of resistance to cisplatin. Therefore, this review is focused on describing mechanisms of cisplatin resistance in ovarian cancer.
Cisplatin
Chemistry and Pharmacokinetics
Cisplatin, chemically cis-diammenedichloro platinum (II) (CDDP), is available in one of three colors – white, yellow, yellow orange. It is crystalline powder at room temperature and is administered intravenously in a saline solution. It binds to albumin and transferrin in blood. 22 Retention half-life of cisplatin is 1-5 -3.6 hours. Further, cisplatin is metabolized by GSH-S-transferase π and conjugates formed in the process are eventually excreted.23
Pharmacodynamics
Cisplatin binds to DNA and forms inter and intra strand cross links, thereby leading to inhibition of DNA synthesis and function.
It is transported into cancer cell by copper membrane transporter (CTR1 & CTR2).24 Once inside the cell, it forms cisplatin-DNA adducts by binding to N7 atoms of guanine or adenine. Later, this results in formation of inter, intra-strand DNA crosslinks and double strand breaks. 9,10 Furthermore, these DNA damages are recognized by proteins like mismatch repair (MMR) proteins, nonhistone high mobility groups 1 and 2 (NHHMG) proteins. 11,25 After recognition of DNA damage, there occurs activation of p53, p73 (pro-apoptotic proteins) pathway, which ultimately induce apoptosis. Cisplatin causes activation ataxia telangiectasia-mutated (ATM) protein and ATM & Rad-3 related (ATR) protein kinase which further induce p53. 25,26 p73 is also a p53 family pro-apoptotic protein. DNA damage caused by cisplatin is recognized by MMR proteins, which in turn activates, Cyclobutane dicarboxylate (c-Abl). Ultimately, c-ABL activates p73.25 Both p53 and p73 pathways activate caspases. Further, caspases can be divided into two types: initiator caspases like caspase 8 & 9, and executioner caspases such as 3 & 7. (Fig.2). 25 These caspases carry out apoptosis. This results in arrest of cell growth in G2/M phase.
Another mechanism of action of cisplatin is generation of superoxide.27 This generation of oxidative stress can result in damage to DNA, protein and lipids, eventually mediate apoptosis.28 Oxidative stress also cause damage to mitochondria by declining state 3 mitochondrial respiration and reducing calcium accumulation. 29 However, these reactive oxygen species are scavenged by glutathione and other antioxidants.
Mitochondrial DNA is also a target for cisplatin action. Cisplatin forms crosslinks with mitochondrial DNA. Moreover, mitochondrial platinum DNA adducts levels are higher than nuclear platinum DNA adducts level. 30 This is due to larger N7 guanine content of the mitochondrial DNA.
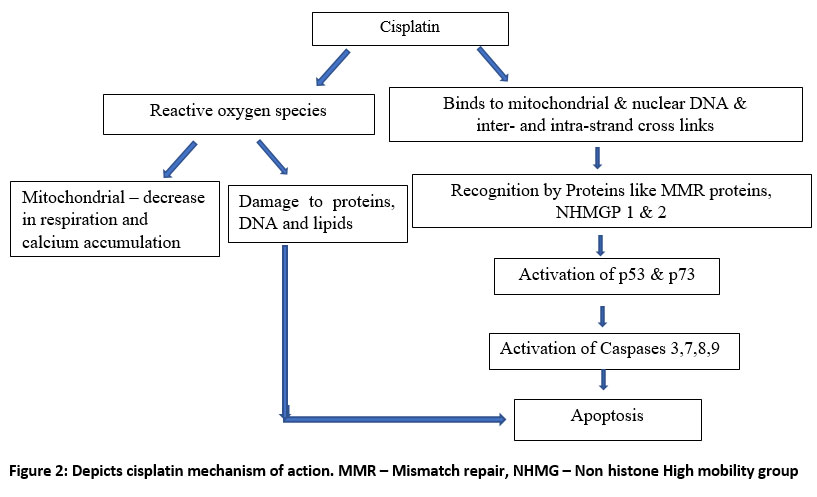 |
Figure 2: Depicts cisplatin mechanism of action. MMR – Mismatch repair, NHMG – Non histone High mobility group |
As described earlier, 50% of HGSOC cases are deficient in homologous recombination (HR), this pathway repairs double stranded breaks in DNA. Cisplatin causes DNA double strand breaks these cannot be repaired in HR deficient cases, thus providing rationale for its use.
Role of cisplatin in other cancer treatments
Small cell lung cancer:- This is an aggressive cancer with elevated potential for metastasis. Majority (70%) of patients are detected at advanced stage. However, those who are detected at early stages are recommended surgical resection along with cisplatin/ carboplatin based chemotherapy. Whereas metastatic disease is incurable, cisplatin/etoposide combination provides symptomatic relief for patients, and recommended as first line therapy. 31 Current guidelines for advanced non-small cell lung cancer , recommend cisplatin based combination chemotherapy. 32
Head and neck squamous carcinoma: It is a common malignant disease. Most of the patients present with locally advanced disease and require surgery followed by chemotherapy and radiotherapy. Cisplatin based combination chemotherapy is preferred in such patients. 33
Other cancers: Cisplatin is used in recurrent brain tumors in pediatric population. It is also used in gastric cancer, anal cancer and leukemia. 25
Mechanisms of cisplatin resistance in ovarian cancer
As mentioned earlier, majority of ovarian cancer patients are detected in advanced stage of disease. Furthermore, these patients develop recurrence after first line of chemotherapy treatment. Recurrent disease is chemo resistant hence fatal to the patient. Therefore understanding the mechanism of development of resistance to overcoming the same is crucial.
Cisplatin resistance is multifactorial and complex. In addition, ovarian cancer cells develop resistance to cisplatin by various mechanisms including conventionally described molecular and tumor microenvironment related mechanisms.
Conventional molecular mechanisms
From conventional perspective, three mechanisms of cisplatin resistance are put forward and those are – reduced intracellular accumulation, increased DNA repair, increased cisplatin metabolism. Table no.1 summarizes these mechanisms.
Reduced accumulation of cisplatin
The most important mechanism of cisplatin resistance is reduced accumulation of cisplatin intracellularly which cause further reduction of cisplatin-DNA adducts formation. Reduction in number of copper transporters 1 & 2,(CTR1 & 2) which transports cisplatin, is seen in ovarian cisplatin resistant cells. 7 Two copper exporters, copper transporting ATPase 7A & 7B (ATP7A & ATP7B) cause sequestration of cisplatin from intracellular components, thus reducing its accumulation.7,23 It is observed that there is increase in expression of these transporters in cisplatin resistant cells. 23
Multidrug resistant proteins (MRP) also play a role in cisplatin resistance. It is seen that increased expression of MRP1 and MRP4 was associated with resistance to cisplatin, with marked reduction in its accumulation.34
Increased cisplatin conversion to inactive metabolite
Cisplatin is metabolized by GSH-S-transferase π and conjugates formed in the process are eventually excreted. 23,35 Increased levels of intracellular glutathione -s- transferase and metallothionine (MT) is seen in cisplatin resistant cells. 36,37 Moreover, in some patients, polymorphism of glutathione S-transferase P1 (GSTP1) Ile105Val gene, results in overexpression of GSH-S-transferase and increased cisplatin inactivation.38 Another study has also found increased expression of long integrated non coding RNA (Linc RNA ) H19, in cisplatin resistant ovarian cancer cells. H 19, is previously known to be involved in tumor development, progression and metastasis. However, this study by Zheng et al, also reveled that H19 increases GSH levels, thereby increasing cisplatin detoxification.39
Increased DNA repair mechanisms
There are four main DNA repair mechanisms and those are, nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), nonhomologous recombination (NHR).23
Nucleotide excision repair (NER)
As mentioned previously, cisplatin acts by forming platinum DNA adducts. NER removes these platinum adducts. This is the major repair mechanism involved in DNA repair. Excision repair cross-complementing 1 (ERCC1) and xeroderma pigmentosum complementation group F (XPF) are proteins involved in carrying out removal of platinum DNA adducts. This is the rate limiting step in the repair mechanism. 8,36,40 However, the increase in expression of ERCC1 is seen in cisplatin resistant ovarian cancer. 41
Mismatch repair (MMR)
This pathway is strand specific and involves in correction of single-base mismatches. This pathway consists of 3 steps – initiation, excision, and re-synthesis and it involves various proteins like MLH1, MSH2, MSH3, MSH6, and PMS2. However, MMR pathway is unable to eliminate DNA-platinum adducts and thus keeping the source of mismatch intact. This will further lead to initiation of MMR pathway and continuation of this futile cycle. This constant procedure of excision and re-synthesis of DNA strand can eventually lead to the formation of double-strand breaks (DSBs). 40As described earlier, this leads to activation of DNA damage signaling factors, including p53, p73, ultimately triggering apoptosis through caspases. Hence unlike other repair mechanisms, deficiency in the MMR pathway can result in cisplatin resistance. This deficiency can occur due to either by epigenetic modification or gene mutation. 40 Moreover, promoter methylation of MMR genes, have shown to cause deficiency of this pathway and thus cisplatin resistance. 41
Homologous recombination (HR)
Cisplatin causes double strand breaks in DNA, which are most dangerous lesions. These lesions can only be repaired by HR and nonhomologous end joining (NHEJ) pathway. HR is a highly conserved system, generally regarded as error-free, that requires an intact sister chromatid to act as template for correct repair of the break without loss of sequence information. As such, HR takes place in G2 and S phases of the cell cycle.
As mentioned previously, proteins encoded by breast cancer gene 1 & 2 (BRCA1 and BRCA2) are responsible to carry out homologous recombination. One such key component is partner and localizer of BRCA2 (PALB2) protein. Mutation in BRCA1 affects binding component capacity of PALB2 and hence compromise homologous recombination. 23 However, study by Da yang has revealed BRCA1 mutation does not confer sensitivity to cisplatin. 42
BRCA2 controls expression of RAD51 protein, which repairs double stranded breaks in DNA. In contrast to BRCA1 mutaions, cells with BRCA2 mutations are incapable of repair and sensitive to cisplatin. 42,43
Nonhomologous end joining pathway (NHEJ) pathway
This pathway also repairs double stranded breaks but unlike homologues recombination, NHEJ is error prone. NHEJ takes place in G1 and G0 phase. Enzymes involved in NHEJ are DNA-dependent protein kinase, DNA polymerase and DNA ligase. Inhibitors of DNA-dependent protein kinase, have shown to increase cisplatin sensitivity, revealing increased expression of this enzyme in cisplatin resistant cells.44
Another mechanism contributing to cisplatin resistance is epigenetic event like alteration in DNA methylation. Ovarian cancer cells which overexpress DNA methyltransferases (DNA-MT) like DNAMT1 & DNAMT3B are known to be cisplatin resistant. 35 Altered methylation of DNA results in chemoresistance. For example hypermethylation mediated suppression of cell adhesion and tight junction, leukocyte trans endothelial migration pathways and DNA hypomethylation mediated activation of cell growth pathways confers cisplatin resistance to cells. 35,45,46
Table 1: Summary of molecular mechanism of cisplatin resistance.
| Regulator | Action mechanism | Effect |
| CTR1 &2 | Decrease in number of | Decrease in influx |
| ATP7A &7B transporters | Increase in number | Increase efflux |
| GSH & MT | Overexpression | Increased cisplatin degradation |
| ERCC1 | Overexpression | Increased DNA repair through NER |
| MMR gene | Promoter methylation | Deficiency of MMR, thus inhibition of apoptosis |
| DNA dependent protein kinase | Overexpression | Increased DNA repair through NHEJ |
| p62, ERK/MAPK, Bcl-2 | Overexpression | Autophagy induction, inhibition of apoptosis |
CTR 1 and 2 – copper transporter 1 and 2, ATP7A & 7B- copper transporting ATPase 7A & 7B, MT- metallothionein, ERCC1 – excision repair cross-complementing 1, NER – nucleotide excision repair, MMR – mismatch repair, NHEJ – Nonhomologous end joining, ERK/MAPK- extracellular regulated kinase/mitogen activated protein kinase, Bcl-2 protein- B cell lymphoma 2 protein.
Tumor microenvironment
Recent studies have shown that tumor microenvironment is involved in tumor progression, metastasis and also chemoresistance. Tumor microenvironment in ovarian cancer is comprised of
Extracellular matrix (ECM), consisting of proteoglycans, glycoproteins, chemokines, cytokines, and metalloproteinases.
Stromal cells like tumor cells, cancer stem cells, cancer associated macrophages and fibroblasts, and cytotoxic T cells.
Blood vessels
Some of the factors related to tumor microenvironment which contributes to cisplatin resistance are as following:
Physical blockade
Two forms of tumor – microenvironment interaction can affect sensitivity to chemotherapeutic drugs: the interaction with soluble factors secreted by stromal cells and direct integration with extracellular matrix (ECM). ECM gene like COL6A3, which codes for collagen VI protein, is upregulated in cisplatin resistant cells.47 This discloses the fact that tumor cells modulate their own environment to protect themselves from cytotoxic drugs like cisplatin.
Tumor microenvironment consist of tightly packed cells leading to poor penetration of drug and drug resistance. 48 Even though molecular mechanisms are main contributing factor for cisplatin resistance, poor penetration through tumor microenvironment do contribute to cisplatin resistance. 48
Hypoxia
Reduced blood supply and tight aggregation of cells results in hypoxia, another contributing factor for cisplatin resistance. Plethora of pathways lead to hypoxia induced resistance, like upregulation of ABC transporter, which results in efflux of cisplatin.49 Furthermore, there is down regulation of several pathways like, MAPK, cell cycle, GAP junction. However, their role in resulting hypoxia induced resistance is yet to be studied. 49 Moreover, hypoxia increases expression of signal transducer and activator of transcription 3 (STAT3) oncogene as well as increase generation of reactive oxygen species. Further, STAT3 expression results in activation of epithelial-mesenchymal transition (EMT), a pathway responsible for cisplatin resistance. 50 Consequences of EMT pathway activation are discussed in detail in later paragraph. Moreover, hypoxia inducible factor -1α (HIF-1α) is involved in imparting chemoresistance to cisplatin. Proposed mechanism is through (HIF-1α) induced autophagy which in turn decreases cisplatin induced apoptosis in cancer cells. 51 In addition, hypoxia promotes cancer stem cells known contributing factor for chemoresistance. 52
Cancer stem cells
As mentioned earlier, cancer stem cells are vital component of tumor microenvironment. They cause tumor growth, metastasis, disease relapse and also imparts chemoresistance to cancer cells. 53 Epithelial-mesenchymal transition (EMT) is cell reprogramming event, occurs in embryonic stages as well as in adult life during tissue injury. In this process, epithelial cells acquire mesenchymal characteristics by losing cell polarity and cell-cell adhesions. They also gain characteristics like invasiveness and migratory potential .54 However, process involves existence of intermediate states including hybrid phenotypes. 55 Tumor cells undergoing EMT and ultimate product of EMT, mesenchymal cells are resistant to cisplatin induced apoptosis. Vimentin, a protein, present in mesenchymal cells is responsible for cisplatin resistance. 56 Furthermore, NANONG, a transcription factor, mediates inheritance of mesenchymal traits leading to chemoresistance in ovarian cancer. It also promotes STAT3 expression, a known booster for EMT pathway and hence chemoresistance. 57 Another promoter of EMT, snail and snug pathway is also found to be upregulated in cisplatin resistant cells. 58
Cancer associated fibroblasts (CAF)
Cancer associated fibroblasts promote tumor progression, metastasis as well as chemoresistance. CAF increases intracellular GSH and cysteine. As described earlier, cisplatin is inactivated by GSH-S-transferase π . Cisplatin-GSH conjugates are eliminated across the membrane through the ATP-dependent glutathione S-conjugate export pump. Hence by increasing GSH, CAF decrease intracellular accumulation of cisplatin. 59 This action of CAF is counteracted by cytototoxic T cells through interferon gamma. 59
Another mechanism by which fibroblasts confer resistance is through secretion of chemokine stromal cell-derived factor-1 (also known as CXCL12). CXCL12 further induce EMT, promoter of chemoresistance, through CXCR4/Wnt/β-catenin pathway. 60–62 Furthermore, CAF promote STAT3 signaling. STAT3 increases expression of antiapoptotic proteins like Bcl-2 and survivin, thus counteracting cisplatin action. 63–64 CAF secrete fibroblast activation protein α (FAP), hallmark of platinum resistance. 65
Microenvironment-Regulated Signaling Pathways
Three pathways PI3K/Akt/mTOR, NFkB, and STAT3 are instrumental in mediating chemoresistance by various mechanisms.
AKT/protein kinase B is an important effector of serine/threonine kinase. It promotes cell survival, suppresses apoptosis, and regulates cisplatin sensitivity in ovarian cancer cell. 66 However, Akt upon phosphorylation inhibits apoptotic proteins like Second mitochondria-derived activator of caspases (Smac), cytochrome c, caspase-9. 66,67
STAT3 pathway is activated by mediators like IL-6, granulocyte-colony stimulating factor (G-CSF), epidermal growth factor (EGF) which are released in tumor microenvironment.63,68 As mentioned earlier, activated of STAT3 further promote inhibitors of apoptosis and EMT. 57,63,64,69 Mediators like IL-6, tumor necrosis factor-α, epidermal growth factors secreted by tumor associated macrophages and dendritic cells activates NFkB pathway. NFkB plays pivotal role in inhibition of cisplatin induced apoptosis.68,70 Besides this, p62 activates NF-κB, and counteract oxidative damage caused by cisplatin. This is through Kelch-like ECH associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor 2 (Nrf2) pathway. 71
Recent advances/research
Autophagy induction: Autophagy is a catabolic pathway in which dysfunctional components, like damaged organelles, misfolded proteins and foreign matter, are transferred into lysosomes for their degeneration. 72 Autophagy and apoptosis are two opposing cell signals which regulate cisplatin sensitivity. In other words, induction of autophagy results in cisplatin resistance. 73 B cell lymphoma 2 (Bcl-2), an antiapoptotic protein, is also an autophagy regulator. BCl-2 associated oncogene (BAG) interacts with Bcl -2 and inhibits cisplatin induced apoptosis. Nevertheless, cisplatin itself promotes BAG and thus confer resistance to ovarian cancer cells. 72 Another study by Juan Wang et al has revealed that cisplatin induces extracellular regulated kinase/mitogen activated protein kinase (ERK/MAP kinase) pathway, which activates autophagy. 74 Moreover, other study showed multifunctional receptor protein, p62, mediates autophagy. 71
As described earlier aberrant methylation of certain genes result in cisplatin resistance. One such gene recently recognized is miR-199a gene. Study by Yuoa Deng, revealed that promoter hypermethylation of miR-199a gene results in overexpression of Discoidin Domain Receptor 1 (DDR 1). DDR1 confers cisplatin resistance as it is involved in regulation of crucial processes like cell proliferation, migration, adhesion and ECM remodeling. 75
Emerging research have proved that noncoding RNAs exert physiological and pathological roles. Noncoding RNAs are further classified into micro RNAs (mi RNAs), long non coding RNAs (lnc-RNAs). Circular RNA (cir RNA) is a novel class of long non coding RNAs. Furthermore, altered expression of non coding RNA including cir RNA is seen in various cancers. In cisplatin resistant ovarian cancer cells 339 cir RNAs are found to be aberrantly expressed. Among these cirRNAs was the cirRNA Cdr1as, whose downregulation was reported in cisplatin resistant ovarian cancer. 76
Micro RNAs (miRNAs), as mentioned earlier, are also non-coding RNAs. Recent research by Pengfei Gen et al, suggested that one such miRNA, mi RNA – 216a is overexpressed in cisplatin resistant cells. Study also elucidated that miRNA -216a downregulates PTEN (Phosphatase Tensin homolog), which is tumor suppressor, thus confering cisplatin resistance to ovarian cancer cells. Furthermore, it was found that miRNA-216a is regulated by STAT3. 77
Overexpression of Nuclear factor erythroid 2 (NF-E2)-related factor 2 (NRF2) is responsible for chemoresistance. Recent study by Lingie Boa and et al, revealed that NRF2 results in activation of ABC (ATP binding cassette) transporters which includes MRPs. These transporters results in enhanced efflux of cisplatin thus causing chemoresistance. 78
Discussion and future prospects
Platinum based chemotherapy is mainstay of ovarian cancer treatment. Large number of patients are diagnosed in advanced stages, because of nonspecific symptoms during early course of disease. However, in advanced stages, development of chemoresistance to cisplatin results in fatal recurrence. One solution suggested for early detection is screening of post-menopausal women either by measuring plasma level of Cancer Antigen 125 (CA125), a biomarker or through ultrasonography. Nonetheless, evidence does not support such screening beneficial for all women. 79 Moreover, such screenings are not feasible in resource poor country like India. Hence way forward is either through use of alternative regimen or combating cisplatin resistance through novel strategy.
Identification of ovarian cancer patients with intrinsically cisplatin resistant tumor is crucial. This can be achieved by identifying biomarkers of cisplatin resistance. Chemoresistance biomarker research is focused mainly on proteomics, transcriptomics and genomics. These include various mi – RNAs, cirRNAs etc. However, use of these biomarkers to detect chemoresistance in clinical practice is not encouraging so far. Furthermore, research and validation of these biomarkers through large group clinical trials will pave way forward. 80
Thorough discussion of mechanism of resistance above, shows that cisplatin resistance is multifactorial and complex. One of the mechanisms is decreased intracellular cisplatin accumulation. Cu transporter, mainly CTR1 is involved in transporting cisplatin intracellularly. Cisplatin competes with Cu for its transport. This discovery have led to research in exploring use of Cu chelators like trientine to counteract cisplatin resistance.81 The clinical benefit rate with the use of tiretine was 33.3 and 50.0% in the platinum-resistant and the partially platinum-sensitive group, respectively.82 However, further Phase II clinical trials are needed to validate the results.
Multi drug resistant proteins which cause increased efflux of cisplatin, are also studied as targets. Metformin has shown to reduce expression of one of the multidrug resistant protein (MRP2) in ovarian cancer cells. 83
Studies have shown resistance mechanisms like increased DNA repair, can be overcome by inhibiting of proteins involved in these processes. Molecules inhibiting ERCC1, a protein involved in nucleotide excision repair, are being studied currently. Also, a inhibitor of replication protein A (catalyzes HR & NER), is being researched. 40
Earlier, these mechanism of resistance like decreased accumulation, increase in DNA repair or increased cisplatin degradation was only known mechanisms. However, recent advances have shown that tumor microenvironment plays an important role in development of cisplatin resistance. Studies undertaken to analyze tumor microenvironment in ovarian cancer have shown tumor cells with the help of other stromal cells modulate environment to their benefit. As discussed earlier, cancer stem cells, cancer associated fibroblast and macrophages do contribute to development of cisplatin resistance. Nevertheless, recent study has also shown cytotoxic T cells present in tumor microenvironment counteract fibroblast mediated cisplatin resistance. 59 Definitely, this should be one of the targets researched, to combat cisplatin resistance in future. Bevacizumab, an antiangiogenic drug, another promising target, attacks tumor microenvironment. Bevacizumab inhibit EMT transition thus counteract cisplatin resistance. 84 Phase III trials demonstrating the safety and efficacy of combining bevacizumab with cisplatin based therapy in advanced stage ovarian cancer are reported. Moreover, results of these trials are promising and showed increase in progression free survival as well as overall survival. 85,86 Few ongoing studies, investigating combination of bevacizumab with immune checkpoint inhibitors are underway. 86
One more novel bioinformatics approach was evaluated to combat cisplatin resistance. This approach found about 535 differentially expressed genes in association of cisplatin resistant ovarian cancer. Furthermore, study also identified sanguinarine, potential inhibitor of epidermal growth factor from natural product library, which can combat cisplatin resistant in ovarian cancer cell lines. 6
ABT737 is an inhibitor of anti-apoptotic protein Bcl-2. As describes earlier, Bcl-2 inhibits cisplatin induced apoptosis. ABT737 by inhibiting Bcl-2 protein found to reverse cisplatin resistance in ovarian cancer cell lines. 87
Alternative strategy devised to target cisplatin resistance is nanotechnology based delivery of cisplatin. This preclinical study showed pegylated nanoparticles encapsulated with cisplatin served as intracellular depot and increased cisplatin efficiency. 86
Additional strategies tested preclinically are employing platinum based prodrug therapy, 88 resensititizing ovarian cancer cells by pretreatment with low dose radiation,89 down regulating hypoxia inducible factor- 1,(90)selective inhibition of tumour associated vacuolar ATPase ‘a2’ isoform to overcome cisplatin resistance in ovarian cancer cells. 90
Combining more than one strategy to overcome cisplatin resistance in ovarian cancer must be future approach.
Acknowledgement
We thank Academic Integrity Committee of Symbiosis Medical College For Women and Symbiosis Centre for Research & Innovation for their support.
Conflict of Interest
There are no conflict of interest.
Funding Source
There are no funding source.
References
- Mathur P, Sathishkumar K, Chaturvedi M, Das P, Sudarshan KL, Santhappan S, et al. Cancer Statistics, 2020: Report From National Cancer Registry Programme, India. JCO Glob Oncol. 2020;(6):1063–75.
CrossRef - Shabir S, Gill PK. Global scenario on ovarian cancer – Its dynamics, relative survival, treatment, and epidemiology. Adesh Univ J Med Sci Res. 2020;2(1):17–25.
CrossRef - Burges A, Schmalfeldt B. Ovarian Cancer Diagnosis and Treatment. Medicine (Baltimore). 2011;108(38):635–41.
CrossRef - Jelovac danijela, Armstrong DK. Recent Progress in the Diagnosis and Treatment of Ovarian Cancer. CA Cancer J Clin. 2011;61(3):183–203.
CrossRef - Robert C. Bast Jr, Bryan Hennessy1 and GBM. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9(6).
CrossRef - Yang L, Zhao H, Yin X, Liang H, Zheng Z, Shen Q, et al. Exploring cisplatin resistance in ovarian cancer through integrated bioinformatics approach and overcoming chemoresistance with sanguinarine. Am J Transl Res. 2020;12(3):923–39.
- Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706–21.
CrossRef - Damia G, Broggini M. Platinum resistance in ovarian cancer: Role of DNA repair. Cancers (Basel). 2019;11(1):1–15.
CrossRef - Makovec T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol Oncol. 2019;53(2):148–58.
CrossRef - Petrović M, Todorović D. Biochemical and molecular mechanisms of action chemical structure of cisplatin mechanisms of action of cisplatin. Med Biol. 2016;18(1):12–8.
- Tanida S, Mizoshita T, Ozeki K, Tsukamoto H, Kamiya T, Kataoka H, et al. Mechanisms of cisplatin-induced apoptosis and of cisplatin sensitivity: Potential of BIN1 to act as a potent predictor of cisplatin sensitivity in gastric cancer treatment. Int J Surg Oncol. 2012;2012(862879).
CrossRef - Momenimovahed Z, Tiznobaik A, Taheri S, Salehiniya H. Ovarian cancer in the world: Epidemiology and risk factors. Int J Womens Health. 2019;11:287–99.
CrossRef - Budiana ING, Angelina M, Pemayun TGA. Ovarian cancer: Pathogenesis and current recommendations for prophylactic surgery. J Turkish-German Gynecol Assoc. 2019;20(1):47–54.
CrossRef - Chen VW, Ruiz B, Killeen JL, Coté TR, Wu XC, Correa CN, et al. Pathology and classification of ovarian tumors. Cancer. 2003;97(10 SUPPL.):2631–42.
CrossRef - Karst AM, Drapkin R. Ovarian Cancer Pathogenesis: A Model in Evolution. J Oncol. 2010;2010:1–13.
CrossRef - Kroeger PT, Drapkin R. Pathogenesis and heterogeneity of ovarian cancer. Curr Opin Obstet Gynecol. 2017;29(1):26–34.
CrossRef - Fathalla MF. Incessant Ovulation-a Factor in Ovarian Neoplasia ? Lancet. 1971;298(7716):163.
CrossRef - Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J Clin. 2019;0(0):1–25.
CrossRef - Chandra A, Pius C, Nabeel M, Nair M, Vishwanatha JK, Ahmad S, et al. Ovarian cancer: Current status and strategies for improving therapeutic outcomes. Cancer Med. 2019;8(16):7018–31.
CrossRef - Book BUYT. Ovarian cancers: Evolving paradigms in research and care. Ovarian Cancers: Evolving Paradigms in Research and Care. 2016. 1–396 p.
- Du Bois A, Lück HJ, Meier W, Adams HP, Möbus V, Costa S, et al. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J Natl Cancer Inst. 2003;95(17):1320–30.
CrossRef - Minami H, Ohe Y, Niho S, Goto K, Ohmatsu H, Kubota K, et al. Comparison of pharmacokinetics and pharmacodynamics of docetaxel and cisplatin in elderly and non-elderly patients: Why is toxicity increased in elderly patients? J Clin Oncol. 2004;22(14):2901–8.
CrossRef - Tapia G, Diaz-Padill I. Molecular Mechanisms of Platinum Resistance in Ovarian Cancer. Ovarian Cancer – A Clinical and Translational Update. 2013.
CrossRef - Ciarimboli G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014;34(1):547–50.
CrossRef - Dasari S, Bernard Tchounwou P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
CrossRef - Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268(10):2764–72.
CrossRef - Masuda H, Tanaka T, Takahama U. Cisplatin generates superoxide anion by interaction with DNA in a cell-free system. Biochem Biophys Res Commun. 1994;203(2):1175–80.
CrossRef - Ozben T. Oxidative stress and apoptosis: Impact on cancer therapy. J Pharm Sci. 2007;96(9):2181–96.
CrossRef - Yuan Yang1, Hong Liu1, Fuyou Liu1,* and ZD. Mitochondrial Dysregulation and Protection in Cisplatin Nephrotoxicity. Arch Toxico. 2014;88(6):1249–56.
CrossRef - Olivero OA, Chang PK, Lopez-Larraza DM, Semino-Mora MC, Poirier MC. Preferential formation and decreased removal of cisplatin-DNA adducts in Chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat Res – Genet Toxicol Environ Mutagen. 1997;391(1–2):79–86.
CrossRef - Farago AF, Keane FK. Current standards for clinical management of small cell lung cancer. Transl Lung Cancer Res. 2018;7(1):69–79.
CrossRef - Sculier JP. Anticancer treatment for advanced non-small cell lung cancer. Breathe. 2011;8(2):125–33.
CrossRef - Rapidis AD, Wolf GT. Immunotherapy of head and neck cancer: Current and future considerations. J Oncol. 2009;2009
CrossRef - Beretta GL, Benedetti V, Cossa G, Assaraf YGA, Bram E, Gatti L, et al. Increased levels and defective glycosylation of MRPs in ovarian carcinoma cells resistant to oxaliplatin. Biochem Pharmacol. 2010;79(8):1108–17.
CrossRef - Li M, Balch C, Montgomery JS, Jeong M, Chung JH, Yan P, et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics. 2009;2:1–13.
CrossRef - Chen SH, Chang JY. New insights into mechanisms of cisplatin resistance: From tumor cell to microenvironment. Int J Mol Sci. 2019;20(17).
CrossRef - Surowiak P, Materna V, Kaplenko I, Spaczyński M, Dietel M, Lage H, et al. Augmented expression of metallothionein and glutathione S-transferase pi as unfavourable prognostic factors in cisplatin-treated ovarian cancer patients. Virchows Arch. 2005;447(3):626–33.
CrossRef - Khrunin A V, Moisseev A. Genetic polymorphisms and the efficacy and toxicity of cisplatin-based chemotherapy in ovarian cancer patients. Pharmacogenomics J. 2010;(September 2009):54–61.
CrossRef - Zheng Z, Xu H, Suo S, Xu X, Ni M, Gu L. The Essential Role of H19 Contributing to Cisplatin Resistance by Regulating Glutathione Metabolism in High-Grade Serous Ovarian Cancer. Nat Publ Gr. 2016;(April):1–12.
CrossRef - Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, Menck CFM. DNA repair pathways and cisplatin resistance: An intimate relationship. Vol. 73, Clinics. 2018. p. 1–10.
CrossRef - Damia G, Imperatori L, Stefanin M, Incalci MD, Farmacologiche R, Negn M. SENSITIVITY OF CHO MUTANT CELL LINES WITH SPECIFIC DEFECTS IN NUCLEOTIDE EXCISION REPAIR TO DIFFERENT ANTI-CANCER AGENTS. Int J Cancer. 1996;66:779–83.
CrossRef - Yang C, Tang D. Association between BRCA2 but not BRCA1 Mutations and Beneficial Survival, Chemotherapy Sensitivity, and Gene Mutator Phenotype in Patients with Ovarian Cancer. J Am Med Assoc. 2011;306(14):1557–65.
CrossRef - Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263–72.
CrossRef - Delacôte F, Lopez BS. Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: The trans-S double-strand break repair model. Cell Cycle. 2008;7(1):33–8.
CrossRef - Chmelarova M, Baranov I, Kovarikova H, Mrkvicova A, Rezacova M, Laco J, et al. Epigenetics & Chromatin. Hered Genet. 2018;07(42):94–5.
CrossRef - Schwarzenbach H, Gahan PB. Resistance to cis- and carboplatin initiated by epigenetic changes in ovarian cancer patients. Cancer Drug Resist. 2019;
CrossRef - Sherman-Baust CA, Weeraratna AT, Rangel LBA, Pizer ES, Cho KR, Schwartz DR, et al. Remodeling of the extracellular matrix through overexpression of collagen VI contributes to cisplatin resistance in ovarian cancer cells. Cancer Cell. 2003;3(4):377–86.
CrossRef - Tannock IF, Lee CM, Tunggal JK, Cowan DSM, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8(3):878–84.
CrossRef - McEvoy LM, O’Toole SA, Spillane CD, Martin CM, Gallagher MF, Stordal B, et al. Identifying novel hypoxia-associated markers of chemoresistance in ovarian cancer. BMC Cancer. 2015;15(1):1–13.
CrossRef - Selvendiran K, Bratasz A, Kuppusamy ML, Tazi MF, Rivera BK, Kuppusamy P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int J Cancer. 2009;125(9):2198–204.
CrossRef - Long F, Liu W, Jia P, Wang H, Jiang G, Wang T. HIF-1α-induced autophagy contributes to cisplatin resistance in ovarian cancer cells. Pharmazie. 2018;73(9):533–6.
- Jalota A, Kumar M, Das BC, Yadav AK, Chosdol K, Sinha S. A drug combination targeting hypoxia induced chemoresistance and stemness in glioma cells. Oncotarget. 2018;9(26):18351–66.
CrossRef - Vinogradov S, Wei X. Cancer stem cells and drug resistance: The potential of nanomedicine. Nanomedicine. 2012;7(4):597–615.
CrossRef - Suster NK, Virant-Klun I. Presence and role of stem cells in ovarian cancer. World J Stem Cells. 2019;11(7):383–97.
CrossRef - Jordan NV, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011;10(17):2865–73.
CrossRef - Li X, Yang J, Wang X, Li X, Liang J, Xing H. Role of TWIST2, E-cadherin and Vimentin in epithelial ovarian carcinogenesis and prognosis and their interaction in cancer progression. Eur J Gynaecol Oncol. 2016;37(1):100–8.
- Qin S, Li Y, Cao X, Du J, Huang X. NANOG regulates epithelial-mesenchymal transition and chemoresistance in ovarian cancer. Biosci Rep. 2017;37(1):1–6.
CrossRef - Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J, et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer. 2012;12.
CrossRef - Wang W, Kryczek I, Dostál L, Lin H, Tan L, Zhao L, et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell. 2016;165(5):1092–105.
CrossRef - Popple A, Durrant LG, Spendlove I, Rolland P, Scott I V., Deen S, et al. The chemokine, CXCL12, is an independent predictor of poor survival in ovarian cancer. Br J Cancer. 2012;106(7):1306–13.
CrossRef - Zhang F, Cui J, Gao H, Yu H, Gao F, Chen J, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Futur Oncol. 2020;16(32).
CrossRef - Mao TL, Fan KF, Liu CL. Targeting the CXCR4/CXCL12 axis in treating epithelial ovarian cancer. Gene Ther. 2017;24(10):621–9.
CrossRef - Wu C-J, Sundararajan V, Sheu B-C, Huang RY-J, Wei L-H. Activation of STAT3 and STAT5 Signaling in Epithelial Ovarian Cancer Progression: Mechanism and Therapeutic Opportunity. Cancers (Basel). 2019;12(24):1–26.
CrossRef - Yan H, Guo BY, Zhang S. Cancer-associated fibroblasts attenuate Cisplatin-induced apoptosis in ovarian cancer cells by promoting STAT3 signaling. Biochem Biophys Res Commun. 2016;470(4):947–54.
CrossRef - Yang Y, Yang Y, Yang J, Zhao X, Wei X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front Cell Dev Biol. 2020;8(August):1–30.
CrossRef - Asselin E, Mills GB, Tsang BK. XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res. 2001;61(5):1862–8.
- Yang X, Fraser M, Moll UM, Basak A, Tsang BK. Akt-mediated cisplatin resistance in ovarian cancer: Modulation of p53 action on caspase-dependent mitochondrial death pathway. Cancer Res. 2006;66(6):3126–36.
CrossRef - Pogge von Strandmann E, Reinartz S, Wager U, Müller R. Tumor–Host Cell Interactions in Ovarian Cancer: Pathways to Therapy Failure. Trends in Cancer. 2017;3(2):137–48.
CrossRef - Johnston PA, Grandis JR. STAT3 signaling: Anticancer strategies and challenges. Mol Interv. 2011;11(1):18–26.
CrossRef - Harrington BS, Annunziata CM. Nf-κb signaling in ovarian cancer. Cancers (Basel). 2019;11(8).
CrossRef - Yan XY, Qu XZ, Xu L, Yu SH, Tian R, Zhong XR, et al. Insight into the role of p62 in the cisplatin resistant mechanisms of ovarian cancer. Cancer Cell Int. 2020;20(1):1–11.
CrossRef - Qiu S, Sun L, Zhang Y, Han S. Downregulation of BAG3 attenuates cisplatin resistance by inhibiting autophagy in human epithelial ovarian cancer cells. Oncol Lett. 2019;18(2):1969–78.
CrossRef - Donovan TRO, Sullivan GCO, Mckenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7(5):509–24.
CrossRef - Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289(24):17163–73.
CrossRef - Deng Y, Zhao F, Hui L, Li X, Zhang D, Lin W, et al. Suppressing miR-199a-3p by promoter methylation contributes to tumor aggressiveness and cisplatin resistance of ovarian cancer through promoting DDR1 expression. J Ovarian Res. 2017;10(1):1–11.
CrossRef - Xu T, Wang M, Jiang L, Ma L, Wan L, Chen Q, et al. CircRNAs in anticancer drug resistance: Recent advances and future potential. Mol Cancer. 2020;19(1):1–20.
CrossRef - Jin P, Liu Y, Wang R. STAT3 regulated miR-216a promotes ovarian cancer proliferation and cisplatin resistance. Biosci Rep. 2018;38(4):1–10.
CrossRef - Bao L, Wu J, Dodson M, Rojo de la Vega EM, Ning Y, Zhang Z, et al. ABCF2, an Nrf2 target gene, contributes to cisplatin resistance in ovarian cancer cells. Mol Carcinog. 2017;56(6):1543–53.
CrossRef - Fields MM, Chevlen E. Ovarian cancer screening: a look at the evidence. Clin J Oncol Nurs. 2006;10(1):77–81.
CrossRef - Pokhriyal R, Hariprasad R, Kumar L, Hariprasad G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark Cancer. 2019;11.
CrossRef - Chen HHW, Kuo MT. Overcoming platinum drug resistance with copper-lowering agents. Anticancer Res. 2013;33(10):4157–62.
- Huang YF, Kuo MT, Liu YS, Cheng YM, Wu PY, Chou CY. A dose escalation study of trientine plus carboplatin and pegylated liposomal doxorubicin in women with a first relapse of epithelial Ovarian, Tubal, and peritoneal cancer within 12 months after platinum-based Chemotherapy. Front Oncol. 2019;9(MAY):1–11.
CrossRef - Du J, Shi H rong, Ren F, Wang J lu, Wu Q hua, Li X, et al. Inhibition of the IGF signaling pathway reverses cisplatin resistance in ovarian cancer cells. BMC Cancer. 2017;17(1).
CrossRef - Aravantinos G, Pectasides D. Bevacizumab in combination with chemotherapy for the treatment of advanced ovarian cancer: A systematic review. J Ovarian Res. 2014;7(1).
CrossRef - Hall M, Bertelli G, Li L, Green C, Chan S, Yeoh CC, et al. Role of front-line bevacizumab in advanced ovarian cancer: The OSCAR study. Int J Gynecol Cancer. 2020;30(2):213–20.
CrossRef - Marchetti C. First-line treatment of women with advanced ovarian cancer: focus on bevacizumab. Onco Targets Ther. 2019;12:1095–103.
CrossRef - Xu Y, Gao W, Zhang Y, Wu S, Liu Y, Deng X, et al. ABT737 reverses cisplatin resistance by targeting glucose metabolism of human ovarian cancer cells. Int J Oncol. 2018;53(3):1055–68.
CrossRef - Wang Z, Deng Z, Zhu G. Emerging platinum(iv) prodrugs to combat cisplatin resistance: From isolated cancer cells to tumor microenvironment. Vol. 48, Dalton Transactions. 2019. p. 2536–44.
CrossRef - Zhao L, Liu S, Liang D, Jiang T, Yan X, Zhao S, et al. Resensitization of cisplatin resistance ovarian cancer cells to cisplatin through pretreatment with low-dose fraction radiation. Cancer Med. 2019;8(5):2442–8.
CrossRef - Ai Z, Lu Y, Qiu S, Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016;373(1):36–44.
CrossRef




