
Manuscript accepted on :09-06-2020
Published online on: 25-06-2020
Plagiarism Check: Yes
Reviewed by: Sharad Kamble
Second Review by: Shabana Khatoon
J.Venkateshwara Rao
Department of Zoology, University College of Science, Osmania University, Hyderabad, Telangana – 500007, India
Corresponding Author E-mail : venbio@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/1946
Abstract
In the present study Human Interleukin IL-29 was produced from human PBMC Cells (Peripheral Blood Mono Nuclear Cells). Upon induction with poly I:C, dendrocytes and PBMC cells produced IL-29. IL-29 molecule is well known to exhibit many anti-proliferative and anti-viral activities in cells and employed as therapeutic agent in many diseases. As its current production is very limited and insufficient, here in this study an attempted to produce it from PBMC cells and expressed in E.coli host. Plasmid vectors were designed with pET-3a vector series with restriction sites NdeI and BamHI and incorporated with IL-29 gene. In the current study the gene was successfully isolated from PBMC cells, m-RNA was extracted and purified converted in to c-DNA by RT-PCR and expressed in host cells. The expressed gene was confirmed on Agarose Gel Electrophoresis and sequences was confirmed with standard gene at NCBI. The study will help for further enhancement and maximise gene expression of IL-29 in host cells and also to scale upto higher levels for industrial scale productions.
Keywords
Antiviral; Anti-Proliferative; Cytokines; Interleukins; PBMC; rhIL-29
Download this article as:| Copy the following to cite this article: Rao J. V. Cloning of Recombinant Human IL-29 (rhIL-29) from PBMC in Escherichia Coli. Biomed Pharmacol J 2020;13(2). |
| Copy the following to cite this URL: Rao J. V. Cloning of Recombinant Human IL-29 (rhIL-29) from PBMC in Escherichia Coli. Biomed Pharmacol J 2020;13(2). Available from: https://bit.ly/3eADw3O |
Introduction
Human interleukin-29 (hIL-29) is a novel cytokine molecule produced from the sources of Peripheral Blood Mono nuclear Cells (PBMC) and mature dendritic cells of human upon viral infection or induction with Poly I:C (Hannig G1,1998). It exhibits anti-proliferative and anti-viral activities through Jak/STAT signalling pathways and up-regulation of the antigen expression of MHC class I (Kotenko, 2003). The O-glycosylated part of glucose portion is not essential for potent action of Homo sapiens IL-29 ( Kang, 1995; Fallah, 2003)as the non-glycosylated portion of recombinant human hIL-29 in E.coli is found to be biologically as active (Komstu, 1987; Delvin 1988; Zhu, 1998 ) as in comparison with expressed in cell lines of mammals (Baneyx,2004).
Hence hIL-29 shoule be produced in E.coli on a large scale at a lower cost (Rosano GL,2014). The human IL-29 gene encoding is derived from human peripheral blood macrophages or tumor cell lines( can be used for cloning in E.coli (Sheppard, 2003). Although the levels of expression is satisfactory but it is not easy express them on industrial scale without vector engineering (Zhipeng Zhou,2016). Hence in the current study vector engineering was done for high expression of gene of interest and also gene was sequenced for confirmation.
Materials and Methods
In the current study plasmid vectors pET-3a, and BL21 (DE3) PLysE were employed. mRNA isolation kit, cDNA synthesis kit, PCR cloning and purification kits, gel extraction kits were purchased from Qiagen Inc, USA. IPTG (Isopropyl β-D-1-thiogalactopyranoside) procured from Genei, Pvt Ltd, Bangalore. Trizol was procured from Sigma, USA. Restriction endonucleases, Chemical reagents and T4 DNA ligases were procured from Invitrogen, USA. Histopaque-1077 supplied from Sigma-Aldrich, USA, Haemocytometer, Inverted Microscope, Leucoseperation centrifuge tubes, CO2 incubator and cell culture facilities utilized from the Dept. of Animal Sciences, University of Hyderabad.
Composition of Media Along with Antibiotics
The PBMC cell culture medium consisted of R.P.MI 1640 which is enriched with heat sterilized fetal calf serum 10%, HEPES buffer 10 mM, L-glutamine 2 mM, 100 U of penicillin per ml, 50 mg of gentamicin per ml and streptomycin 100 mg per ml.
PBMC Cells Isolation from Human
Human PBMC (Peripheral blood Mononuclear cells) were extracted from thick solid buffy coats, which were separated as fraction of a centrifuged blood sample which holds more quantity of leukocytes. LeucoSep centrifuge tubes employed to isolate leucocytes with 15 mL Ficoll-Histopaque solution and done centrifugation at 250g for 15 min.
Isolation of Total RNA
Mononuclear cells isolated by Ficoll-Histopaque method were grown in R.P.MI 1640 media [10% of Fetal bovine serum Pencillin ( 100 I.Units per ml), Gentamycin (100 μg per ml), Streptomycin (100 μg per ml) and lectin (125 μg per ml) in CO2 incubator with 5% CO2 at 37 0C for 48 hours. In the growing PBMC cells interleukin genes were induced with (poly I:C) Polyinosinic: polycytidylic acid (50 μg per ml) and incubated for 48 hours. Cells were suspended in Trypsin-EDTA buffer and separated at 1500g for 10 min.
The obtained cell pellet was kept in 1 ml of Trizol and kept for 5 minutes on ice. Chloroform of 200 μl was added to tubes & after vortexing for 15 sec, incubated for 15 minutes on ice. Then lysate was separated by 12000 g for 15 minutes at 40 0C and in a separate tube supernatant was collected. RNA content of supernatant was precipitated with Isopropanol and purified with 75% ethanol wash. The alcohol free pellet of RNA was mixed in MilliQ nuclease free water and at 60 0C incubated for 10 minutes. Then, to check purity 5 μl of sample was resolved on 1% agarose gel.
m-RNA Isolation
About 250 μl of total RNA isolated was taken into 15ml FastTrack Lysis Buffer (containing mixture of RNAse inhibitors) and mixed thoroughly. The reaction complex was heated for 5 minutes toat 650 C and then placed for 1 minute on ice. To this, 5M NaCl of 950 μl of was added and thoroughly mixed by swirling. Oligo dT column washed twice with 50 mM sodium citrate, 0.5 M LiCl and 0.1% SDS (binding buffer), then sample was passed through Oligo dT column and allowed to settle for 15 minutes. This was washed twice with 10 ml Binding Buffer. The mixture was incubated at RT for 3 to 4 hours and centrifuged at 3000g for 5 min. 100 μl mRNA was eluted from Oligo-dT column using 0.1% SDS, 1 mM sodium citrate [elution buffer] & elute was separated into a fresh 2ml Eppendorf tube.
RT-PCR and Synthesis of cDNA
About 2μg of total RNA was used for reverse transcription experiment. It was executed at 47 0C for a period of 30 min and cDNA amplification was done by using one step kit of RT-PCR (Qiagen). Primers were designed by using Fast PCR software for the complete-length fragment of human IL-29 gene including BamH1 and Nde1 restriction sites (indicated by underlines).
The primers and sequences are as follows: `GCGCCATATGGGCCCTGTCCCAACTTCC` used as forward primer and `GCGGATCCTCAGGTGGACTCAGGGTGGGTTG` as reverse primer. RNA sample, dNTP mix, Onestep RT-PCR buffer, primer solutions 2 x Qiagen, and RNase free water was initially kept for 15 min on ice. For master mix working volume of 50 μl was prepared with the following ingredients: 10 μl 1X Qiagen, One-step RT- PCR buffer, 2 μl of Forward and reverse primers with 0.6 μM, 2 μl of 400 μM of each of dNTP’s, RNase inhibitor of 5 units, template RNA of 2 μg and 2 μl of Qiagen RT-PCR enzyme mix. In PCR tubes this preparation was placed and mixed gently. RT PCR cycles were operated as below: Firstly , Reverse Transcription held for 30 minutes at 50 OC, Initial PCR pre activation step at 95 OC held for 10 min during which hot star Taq DNA polymerase was activated, immediately followed by 40 cycles of 3-step PCR: Denaturation was done at 94 OC done for 1 min, Annealing executed at 65 OC for 1 min and Extension was done at 72 OC for 2 min followed by final extension performed at 72 OC for 10 min. Finally by using Qiagen PCR purification kit, the PCR product was purified. Then on 1% agarose gel PCR product was analyzed.
Plasmid Isolation
A single colony of bacterial culture containing pET 3a was transferred with 20 ml of LB broth with Ampicillin (50 μg per ml) & incubated over-night at 370 C. 6 ml of each culture was harvested in eppendorf tubes and then pellet was held with 150μl of TEG [Tris-EDTA-Glucose; Solution 1). To this 250 μl of lysis solution (1M NaOH with 10% SDS) was added by gentle mixing and after kept it on ice for 3 minutes. Ice cold 3M Sodium acetate of 300 μl was added and after gentle mixing by toppling, kept for 10 min on ice at 4 0C. Then, the tubes were centrifuged at 12000 g for 10 min at 40C and supernatant was collected into a separate tube without disturbing the precipitated proteins. The DNA plasmid in the supernatant was further purified by phenol chloroform precipitation and 70% ethanol wash. After removal of residual alcohol plasmid DNA was held in 50 μl of milliQ water. Isolated plasmid of 5 μl loaded with 1% agarose gel and resolved on gel.
Restriction Digestion and Gel Elution
Restriction digestion of product PCR and plasmid was carried by taking 10X restriction enzyme buffer of 2 μl, 2 μg of DNA dissolved in MilliQ water to make a final volumes of 18 μl. Each 1 µl (10 units) of BamH I and Nde I added and mixed gently. Then reaction mixture was centrifuged for a few seconds in a microfuge and incubated at 370 C for 2 hours. A small aliquot was resolved on a gel to check the digestion. After restriction digestion DNA was resolved on 1% low melting agarose gel. Under low voltage UV, the desired band was excised using a sterile blade. The excised gel slice taken in a eppendorf tube of 1.5 ml for 15 minutes and kept at -700 C. Slices were melted by incubating the tube at 650 C. After adding equal-volumes of TE-saturated phenol and vortexing for 30 seconds, the sample was kept at -700 C for 30 minutes. The sample was thawed & centrifuged in a micro centrifuge at 10,000 r.p.m for 5 minutes at room temp to sort out the phases. Carefully the aqueous phase was removed in to a clean tube. Then 1/10th volume Isopropanol was employed for DNA precipitation and with ethanol DNA pellet dried. The DNA purity inspected on 1% conc. of agarose gel electrophoresis.
Construction of Recombinant Expression Vector and Transformation
Standard DNA tmolecular techniques utilized for building of the plasmid recombinant vectors [Sambrook et.al, 2001]. Manual alkali lysis method was employed for isolation of plasmid DNA. The PCR amplified gene (546bp) and pET-3a vector were digested with restriction enzymes Nde1 and BamH1, then fragments were extracted by using nucleic acid purification column [Qiagen) and ligated with T4 DNA ligase in the proportion of 1:5 with overnight incubation at 160C. By CaCl2 method, Competent cells of BL21 (DE3) PLysS were prepared and transformed with recombinant vector pET-3a containing the IL-29 gene of human and plated on LB agar containing 25 mg/mL & Chloramphenicol 50 mg/mL of Ampicillin. Transformants screened out by using antibiotic selection markers.
Confirmation of Recombinants
Confirmations of recombinants of rhIL-29 gene were done by Restriction Digestion of plasmids followed by gel analysis, PCR and Sequencing Methods.
A) Restriction Digestion
Restriction digestion of PCR product and plasmid was carried out with NdeI and Bam HI restriction enzymes at 370 C for 2 hours. A small aliquot was resolved on a gel to check for digestion.
B) PCR
Primers were designed for IL-29 using softwareprogram Fast-PCR server located at http://www.biocenter.helsinki.fi/bi/Programs/fastpcr. Forward Primer `GCGCCATATGGGCCCTGTCCCA ACTTCC` and reverse primer `GCGGATCCTCAGGTGGACTCAGGGTG GGTTG` were employed to amplify hIL-29 gene using PCR. PCR was performed employing the following parameters; 95 0C for about 2 min, 20 cycles kept 95 0C for 60 sec, 50 0C for hold for 60 sec, 70 0C for keeping 2 min and 70 0C for holding 10 min after last cycle. The product of PCR was then analyzed on 1% agarose gel.
C) Sequencing
Recombinant clone of rhIL-29 was sequenced with the same forward (`GCGCCATATGGGCCCTGTCCCAACTTCC`) and reverse primers (`GCGGATCCTCAGGTGGACTC AGGGTGGGTTG`) that were specific for IL-29 using an automated sequencer ABI PRISM 3100 Genetic analyzer and the obtained data was compared with GenBank database employing the BLAST software browsed at NCBI web server addressed at (www.ncbi.nlm.nih.gov/BLAST).
Results and Discussion
The PBMC (Peripheral blood mono nuclear cells) which were collected using Histopaque 1077 were observed under the microscope. Live and healthy cells were identified by using Tryphan blue stain. More than 85% of cells were found healthy (Table 1). The cell number was counted using haemocytometer. The count was found to be 2.4 x 106 cells per ml. The healthy cells were carefully separated by low speed centrifugation at 2000 r.p.m, then induced with poly I: C (50 μg/ml) and incubated with 5% CO2 in CO2 incubator for 48 hrs.
 |
Figure 1:PBMC cell -Under Microscope observation |
After 48 hrs of post poly I:C induction of PBMC (Fig 1), total RNA was collected as described in methods. The quality of RNA was checked by taking O.D at 260 and 280 nm respectively and the ratio was found to be 1.99. In first four lanes fresh total RNA sample (8 µl) was loaded. Fifth lane corresponds to the negative control and in sixth lane sample after 24 hours of incubation at 40C was loaded. In gel, lower band corresponds to 18S RNA and the upper band corresponds to 23 S RNA respectively. Two clear bands [28S, 18S RNA] resolved on 1% agarose gel indicating high purity of RNA in the sample. mRNA eluted from oligodT column was quantitated by Orcinol method has given a yield of 50 μg/ml. About 0.5 μl of mRNA collected from the above step was utilized in performing RT-PCR with IL-29 specific primers. A single amplicon of about ~550 bp was obtained which was almost the size equal to IL-29, indicating the success of RT-PCR. After NdeI and BamHI restriction digestion, the amplicon and plasmid were resolved on 1% agarose gel. The cut plasmid DNA was resolved at 4.3 Kb and the amplicon was resolved at ~550 bp as an individual single band. The bands of interest were sliced from gel and the DNA was eluted using gel elution kit (Qiagen). The quality of DNA was observed on 1% agarose gel. Purified DNA was found to be pure and containing 150 μg/ml amplicon and 200 μg/ml cut plasmid.
The human IL-29 cDNA encoding gene was cloned (Fig. 2) as described. The 546 bp IL-29 gene was cloned under T7 promoter in pET 3a vector. At the end a transcriptional terminator was taken. The nucleotide sequence of the selected IL-29 was homologous to that of native sequence deposited with Genbank (NM_172140). Using standard laboratory protocols the rhIL-29 expression was studied and found to be very low (65 mg/L). So, further studies were initiated to improvise the expression of rhIL-29 by using codon substitution methods and also optimization of culture conditions.
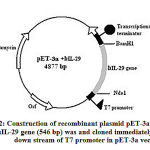 |
Figure 2: Construction of recombinant plasmid pET-3a + IL-29. |
The constructed recombinant plasmid was confirmed by resolving 1% agarose gel. The original plasmid pET 3a showed corresponding band at 4.3 Kb (Lane No.2) where as recombinant plasmid showed corresponding band at ~5.3 Kb. (Lane No.3) in Fig 3.
Confirmation of Recombinant Plasmid (pET 3a + IL-29)
The constructed recombinant plasmid was confirmed by A) Restriction digestion. B] PCR and c] Sequencing. The plasmid isolated from positive clones was used for confirmation.
A) Restriction Digestion
Recombinant plasmid isolated from positive clone was subjected to restriction digestion with BamH I and Nde I. Recombinant plasmid and digested recombinant plasmid were loaded on 1% agarose gel. Recombinant plasmid was resolved at 4.8 K.bp whereas digested plasmid showed 4.3 and ~0.6 K.bp bands, indicating the successful insertion of IL-29 gene in pET plasmid.
B) PCR
The plasmid was extracted from selected positive clones and used for PCR amplification of IL-29. A single band of ~540 bp was obtained confirming the presence of IL-29 in the recombinant plasmid.
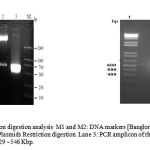 |
Figure 3: PCR and restriction digestion analysis M1 and M2 |
C) Sequencing
The IL-29 gene in pET 3a vector was sequenced with ABI Prism 342 sequencer using `GCGCCATATGGGA CCTGTTCCAACTTCCAAGCCAACCAC as forward primer and GCGGATCCTCAGGTGGACT CAGGGTGGGTTG` as reverse primer. The sequenced information found well-matched with IL-29 gene sequence present in the database of Genbank [NCBI).
Conclusion
In this study we attempted to produce IL29 from PBMC cells and expressed in E.coli. Plasmid vectors were designed with pET-3a series vectors and incorporated with IL-29 gene. The IL-29 gene was successfully expressed from PBMC, m-RNA was isolated and purified converted to c-DNA by RT-PCR and cloned in host cells. The expressed gene was confirmed on Agarose Gel Electrophoresis and PCR methods. This study can help for further optimization step of gene expression of IL-29 in E.coli and also to scale-up to industrial level production.
Acknowledgement
The author is highly thankful University of Hyderabad and JNTU, Hyderabad for providing research facilities.
Conflict of Interest
There is not funding source
References
- Kotenko SV et.al.,(2003). IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003 Jan;4(1):69-77. Epub 2002 Dec 16.
- Sheppard P1 et,al., (2003). IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003 Jan;4(1):63-8. Epub 2002 Dec 2.
- Baneyx F1 (2004) Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004 Nov;22(11):1399-408.
- Hannig G1, Makrides SC (1998). Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 1998 Feb;16(2):54-60.
- Rosano GL, Ceccarelli EA (2014). Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014;5:172. Published 2014 Apr 17. oi:10.3389/fmicb.2014.00172
- Zhipeng Zhou, Yunkun Dang (2016) The role of codon usage on gene expression
- Proceedings of the National Academy of Sciences Oct 2016, 113 (41) E6117-E6125; DOI: 10.1073/pnas.1606724113
- Komastu, Y., et.al., (1987). cloning of granulocyte colony stimulating factor cDNA and its expression in Escherichia coli. Japanese Journal of Cancer Research, 78, 1179–1181
- Devlin PE et.al., (1988) Alteration of amino-terminal codons of human granulocyte-colony-stimulating factor increases expression levels and allows efficient processing by methionine aminopeptidase in Escherichia coli. Gene 65:13–22
- Shu, Z. H., & Qinong, Y. et.al., (1998). Expression of cDNA for rhG-CSF in E. coli and characterization of the protein. Chinese Journal of Cancer Research, 10, 256–259.
- Kang, S. H. et.al., (1995). High-level expression and simple purification of recombinant human granulocyte colony stimulating factor in E. coli. Biotechnology Letter, 17, 687–692.
- Fallah, M. J., et.al.,(2003). Over expression of recombinant human granulocyte colony stimulating factor in E. coli. I J M S, 28, 131–134.
- Sambrook J, Russell D (2001) Molecular Cloning: A Laboratory Manual, 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.

