
Manuscript accepted on :23-May-19
Published online on: 18-06-2019
Plagiarism Check: Yes
Reviewed by: Ankur Singh Bist
Second Review by: Dini Damayanti
N. N. Bahari1 , S. Y. N. Jamaludin1*, A. H. Jahidin2, M. N. Zahary3 and A. B. Mohd Hilmi3
, S. Y. N. Jamaludin1*, A. H. Jahidin2, M. N. Zahary3 and A. B. Mohd Hilmi3
1Faculty of Medicine, Universiti Sultan Zainal Abidin, Medical Campus, Terengganu, Malaysia.
2Faculty of Pharmacy, Universiti Teknologi MARA, Puncak Alam Campus, Selangor, Malaysia.
3Faculty of Health Sciences, Universiti Sultan Zainal Abidin, Gong Badak Campus, Terengganu, Malaysia.
Corresponding Author E-mail: yusrinanadihah@unisza.edu.my
DOI : https://dx.doi.org/10.13005/bpj/1683
Abstract
The transient receptor potential vanilloid member 4 (TRPV4) is a non-selective calcium (Ca2+)-permeable channel which is widely expressed in different types of tissues including the lungs, liver, kidneys and salivary gland. TRPV4 has been shown to serve as a cellular sensor where it is involved in processes such as osmoregulation, cell volume regulation and thermoregulation. Emerging evidence suggests that TRPV4 also plays important roles in several aspects of cancer progression. Despite the reported roles of TRPV4 in several forms of cancers, the role of TRPV4 in human colorectal cancer remains largely unexplored. In the present study, we sought to establish the potential role of TRPV4 in colorectal cancer by assessing TRPV4 expression levels and investigating whether TRPV4 pharmacological modulation may alter cell proliferation, cell cycle and cell death in colorectal cancer cells. Quantitative real-time PCR analysis revealed that TRPV4 mRNA levels were significantly lower in HT-29 cells than normal colon CCD-18Co cells. However, TRPV4 mRNA was absent in HCT-116 cells. Pharmacological activation of TRPV4 with GSK1016790A significantly enhanced the proliferation of HT-29 cells while TRPV4 inhibition using RN 1734 decreased their proliferation. Increased proliferation in GSK1016790A-treated HT-29 cells was attenuated by co-treatment with RN 1734. Pharmacological modulation of TRPV4 had no effect on the cell cycle progression but promoted cell death in HT-29 cells. Taken together, these findings suggest differential TRPV4 expression levels in human colorectal cancer cells and that pharmacological modulation of TRPV4 produces distinct effects on the proliferation and induces cell death in HT-29 cells.
Keywords
Cell Cycle; Cell Death; Colorectal Cancer; Proliferation; TRPV4
Download this article as:| Copy the following to cite this article: Bahari N. N, Jamaludin S. Y. N, Jahidin A. H, Zahary M. N, Mohd Hilmi A. B. Assessment of TRPV4 Channel and Its Role in Colorectal Cancer Cells. Biomed Pharmacol J 2019;12(2). |
| Copy the following to cite this URL: Bahari N. N, Jamaludin S. Y. N, Jahidin A. H, Zahary M. N, Mohd Hilmi A. B. Assessment of TRPV4 Channel and Its Role in Colorectal Cancer Cells. Biomed Pharmacol J 2019;12(2). Available from: https://bit.ly/2RlWkc9 |
Introduction
According to the GLOBOCAN 2018 estimates, colorectal cancer (CRC) remains as one of the dominating types of cancer, responsible for over 1.8 million new cancer cases and 881,000 deaths worldwide, ranking in third place for cancer incidence and second place for mortality.1 In addition to the well-established role of genetic factors in the pathogenesis of CRC,2 it is also recognised that several ion channels have been implicated in the process of developing CRC.3-5 Indeed, ion channels have been reported to serve as potential biomarkers in several types of cancer including prostate, breast, lung, pancreas, esophageal and colorectal cancer.6 Among these ion channels, several members of the transient receptor potential (TRP) channels have been identified as promising cancer biomarkers,6 such as TRP canonical 6 (TRPC6) where it is upregulated in esophageal cancer and is associated with poor prognosis.7
The mammalian TRP channel superfamily comprises six subfamilies: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin) and TRPA (ankyrin).8 In general, TRP channels function as sensors in various types of cells and they are also responsible for providing route of Ca2+ entry to regulate important Ca2+-sensitive processes such as gene transcription and cell death.8 Of the six members in the TRPV subfamily (TRPV1-6),8 TRPV4 has attracted a great interest because of its link to several human diseases9 and its therapeutic potential in diseases of the digestive system,10 neuropathic pain,11 obesity and type 2 diabetes.12 TRPV4 is a non-selective Ca2+ permeable cation channel found to be widely expressed in many types of tissues including the brain, heart, liver and lungs.13 Previous studies have provided evidence for the critical role of TRPV4 in various cellular functions such as thermoregulation, mechanosensation and osmoregulation.14-16
Recent studies have also implicated TRPV4 in some types of cancer. A study by Lee et al.,17 implicated TRPV4 in breast cancer metastasis. The authors discovered that TRPV4 expression is enhanced in basal subtype of breast cancer and that inhibition of TRPV4 (either using an inhibitor or TRPV4 silencing) in breast cancer cells impaired their migration and invasion, indicating that TRPV4 is involved in metastasis.17 The pro-migratory and pro-metastatic effects of TRPV4 in breast cancer appear to be mediated by Ca2+-dependent activation of AKT and downregulation of E-cadherin cell cortex protein.18 Another recent study by Peters et al.,19 has provided evidence for the role of TRPV4 in promoting breast cancer cell death, particularly in those cells with high endogenous TRPV4 expression levels. The authors observed that TRPV4 activation using GSK1016790A decreased the viability of MDA-MB-468 and HCC1569 basal breast cancer cells which expressed high levels of TRPV4. Further experiments uncovered that TRPV4 activation produces cell death in MDA-MB-468 breast cancer cells via oncosis and apoptosis. Using an orthotopic mouse model, they also showed that TRPV4 activation decreased tumour growth in vivo, further highlighting the potential utility of TRPV4 as a therapeutic target for breast cancers which overexpress TRPV4 channel.19
Unlike breast cancer, studies assessing TRPV4 in the context of colorectal cancer have not been extensive. Previous studies have suggested a role for TRPV4 in the proliferation of colorectal cancer cells. Wasilewski et al.,20 found that the viability of human colon adenocarcinoma Colo-205 cells was markedly reduced in response to co-incubation of fatty acid amide hydrolase inhibitor PF-3845 with the TRPV4 antagonist RN 1734,20 indicating that TRPV4 may be involved in regulating the proliferation of Colo-205 cells. However, additional investigations are required to further corroborate this finding given that their studies did not include the assessment of TRPV4 expression levels in Colo-205 cells. Another study by Sozucan et al.,21 has identified that TRPV4 is expressed at a low level in the CRC patients tissue samples compared with the normal tissues. Their studies however, did not explore the possible mechanism to explain the observed downregulation of TRPV4 in colorectal cancer, although they did postulate that epigenetic factors may contribute to the suppression of TRPV4 expression.21
Owing to the lack of studies assessing the role of TRPV4 particularly in colorectal cancer,22 the present study investigated the possible involvement of TRPV4 in regulating cancer-related events. In the present study, we aimed to determine TRPV4 expression in two human colorectal cancer cell lines and assess its role in regulating the proliferation, cell cycle and cell death in these cells.
Materials and Method
Cell Culture
The human colorectal cancer cell lines HT-29 (ATCC HTB-38) and HCT-116 (ATCC CCL-247) were maintained in RPMI-1640 (Gibco) culture medium supplemented with 10% foetal bovine serum (FBS) (Gibco), penicillin 100 U/mL and streptomycin 100 µg/mL (Gibco). Fibroblasts from normal colon (CCD-18Co, ATCC CRL-1459) were cultured in Eagle’s Minimum Essential Medium (EMEM) (ATCC) supplemented with 10% FBS (Gibco), penicillin 100 U/mL and streptomycin 100 µg/mL (Gibco). All cells were cultured in a humidified incubator (37°C, 5% CO2) and routinely observed for any morphological changes or contamination.
RNA Isolation and Quantitative real-time PCR
RNA was isolated from HT-29, HCT-116 and CCD-18Co cells (from three different biological replicates) using RNeasy Plus Mini Kit (Qiagen) by following the manufacturer’s protocol. Total RNA from each sample was reverse-transcribed by using Omniscript Reverse Transcriptase kit (Qiagen) with random primers and RNase inhibitor (Promega) according to the manufacturer’s instructions. Samples were incubated for 60 minutes at 37°C in a T100™ Thermal Cycler (BioRad). Subsequent quantitative real-time PCR was performed by using Assays-on-Demand primer/probe sets and TaqMan Universal PCR Master Mix (Applied Biosystems). Assays included TRPV4 (Hs01099348_m1, FAM-MGB primer/probe) and the reference gene 18S ribosomal RNA (4319413E, VIC-MGB primer/probe). All amplifications were done using the following thermal cycling conditions: 10 minutes at 95°C (holding stage), followed by 40 cycles of denaturation for 15 seconds at 95°C and combined annealing and extension steps for 1 minute at 60°C in a StepOnePlus™ Real-Time PCR System Thermal Cycling Block (Applied Biosystems). Gene expression data were normalised to 18S ribosomal RNA (4319413E, Applied Biosystems) and calculated using the comparative Cq method as previously described (23).
MTT Cell Proliferation Assay
HT-29 cells (1 x 104 cells per well) were seeded in 96-well plates. At 24 hour post-seeding, the cells were treated with DMSO (vehicle), GSK1016790A (Sigma-Aldrich) (1-100 nM) or RN 1734 (Tocris Bioscience) (1-100 µM). For combination drugs treatment, 100 nM of GSK1016790A and 10 µM of RN 1734 were used. The final DMSO concentration in each well for all treatment groups was maintained between 0.1% and 0.11% throughout the experiments. At 72 hours post-treatment, an MTT cell proliferation assay (Sigma-Aldrich) was carried out by adding 20 µL of MTT stock solution (5 mg/mL) into each well. The plates were incubated for 4 hours at 37ºC. Following incubation, the MTT-containing medium was discarded and 150 µL of DMSO was added to dissolve the formazan crystals. The absorbance (Abs) in each well was measured at 570 nm by using a microplate reader (Tecan Infinite M200 Pro). The cell proliferation (viability) was determined by using the following formula:

Cell Cycle Analysis
Assessment of the cellular DNA content was performed by cell cycle analysis using flow cytometry. HT-29 cells were seeded in 6-well plates at 5 x 105 cells per well (24). At 24 hour post-seeding, the cells were treated with 0.11% DMSO (vehicle), GSK1016790A (Sigma-Aldrich) (100 nM), RN 1734 (Tocris Bioscience) (10 µM) or co-treatment with GSK1016790A and RN 1734 (100 nM and 10 µM, respectively). Cell cycle analysis was performed at the time points stated in the results by using BD Cycletest™ Plus DNA Kit (BD Biosciences). Briefly, cells were harvested after trypsinisation and centrifugation at 1780 rpm for 5 minutes. The cell pellet obtained were fixed in 70% cold ethanol and stored at 4°C. The fixed cells were then stained with 200 µL propidium iodide (PI) stain solution (BD Biosciences) prior to analysis with CytoFLEX flow cytometer (Beckman Coulter). An amount of 10 000 cells was analysed for each sample.
Annexin V/PI double-staining Assay
HT-29 cells were plated at 5 x 105 cells per well in 6-well plates and incubated overnight (24). At 24 hour post-plating, the cells were treated with 0.11% DMSO (vehicle), GSK1016790A (Sigma-Aldrich) (100 nM), RN 1734 (Tocris Bioscience) (10 µM) or co-treatment with GSK1016790A and RN 1734 (100 nM and 10 µM, respectively). Heat-induced cells (i.e. 2 minutes incubation at 60°C) were included as a positive control for this assay. Assessment of cell death was performed at the time points indicated in the results by using BD Pharmingen™ FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) in accordance with the manufacturer’s guidelines. In brief, the cells were washed with cold phosphate buffered saline and detached by enzymatic trypsinisation (25) using TrypLE (Gibco). The cells were centrifuged at 1780 rpm for 5 minutes and the cell pellet was then resuspended in 1 x binding buffer. An amount of 5 µL of FITC-Annexin V and 5 µL of PI was added to 100 µL of the cell suspension containing 105 cells. After gently vortexing, the cell suspension was incubated at room temperature for 15 minutes in the dark. Following incubation, 400 µL of 1 x binding buffer was added and assessment of cell death was performed using CytoFLEX flow cytometer (Beckman Coulter). An amount of 10 000 cells was analysed for each sample.
Statistical Analysis
All statistical analyses were done using IBM SPSS Statistics or GraphPad Prism. The statistical tests used are indicated in each figure legend. Results are expressed as mean ± standard deviation (SD). P < 0.05 was considered as statistically significant.
Results
TRPV4 is downregulated in human colorectal cancer cell lines
We examined the relative expression of TRPV4 mRNA levels in two different human colorectal cancer cell lines; HT-29 and HCT-116, and compared with the non-transformed normal colon, CCD-18Co cells. We observed that TRPV4 mRNA levels were significantly lower in HT-29 cells than the normal colon, CCD-18Co cells (Figure 1). In HCT-116 cells however, TRPV4 mRNA expression was undetected (Figure 1).
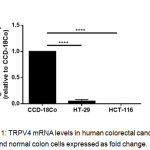 |
Figure 1: TRPV4 mRNA levels in human colorectal cancer cell lines and normal colon cells expressed as fold change.
|
Data are presented as mean ± standard deviation (n=3) from three independent cell passages. Statistical analysis was performed using an unpaired t-test, **** P < 0.0001.
Pharmacological modulation of TRPV4 produces opposing effects on the proliferation of HT-29 colorectal cancer cell lines
To assess the possible role of remaining TRPV4 in CRC and its potential for therapeutic targeting, the effect of TRPV4 pharmacological modulation on the proliferation of HT-29 cells was investigated. Using an MTT cell proliferation assay, we found that treatment with a selective TRPV4 activator GSK1016790A (i.e. 10 nM and 100 nM) increased the proliferation of HT-29 cells (Figure 2A). By contrast, pharmacological inhibition of TRPV4 using RN 1734 (i.e. 10 µM and 100 µM) reduced the proliferation of this cell line (Figure 2B). GSK1016790A-mediated increases in proliferation of HT-29 cells was prevented by co-treatment with RN 1734 (Figure 2C).
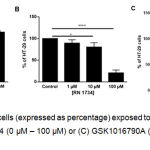 |
Figure 2: Viability of HT-29 cells (expressed as percentage) exposed to (A) GSK1016790A (0 nM – 100 nM), (B) RN 1734 (0 µM – 100 µM) or (C) GSK1016790A (0.1 µM) and RN 1734 (10 µM) for 72 hours.
|
Data are presented as mean ± standard deviation (n=3-4). Statistical analysis was performed using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test for (A) and (B) or Tukey’s multiple comparisons test for (C), *P < 0.05, **P < 0.01, *** P < 0.001, **** P < 0.0001.
Pharmacological modulation of TRPV4 does not alter the cell cycle progression of HT-29 colorectal cancer cell lines
To further explore the potential role of TRPV4 in colorectal cancer cells, we assessed the effect of TRPV4 pharmacological modulators on the cell cycle progression of HT-29 cells, both at 24 and 48 hours (Figure 3). The FACS analysis demonstrated that the highest percentage of cells were in G0/G1-phase for all treatment groups, which was not significantly different from the DMSO control (Figure 3B and 3D). Similarly, no difference was observed in both S- and G2/M-phases for all treatment groups compared with the DMSO control. Taken together, this observation suggests that pharmacological modulation of TRPV4 had no profound effects on the cell cycle profile in HT-29 cells.
 |
Figure 3: Cell cycle analysis of DNA contents in HT-29 cells.
|
Representative DNA histogram showing the cell cycle phase distribution of control and treated cells at 24 hours (A) and 48 hours (C). Bar graphs indicate the quantified percentage of HT-29 cells in each cell cycle phase at 24 hours (B) and 48 hours (D). Data are presented as mean ± standard deviation (n=3). Statistical analysis was performed using two-way ANOVA with Bonferroni multiple comparisons test.
Pharmacological modulation of TRPV4 promotes cell death in HT-29 colorectal cancer cell lines
Next, we examined whether or not pharmacological modulation of TRPV4 could induce apoptosis in HT-29 cells. Using Annexin-V/PI double-staining assay (Figure 4), we observed that treatment with GSK1016790A alone induced apoptosis in HT-29 cells in a time-dependent manner, as depicted by the highest percentage of apoptotic cells compared with the DMSO control (Figure 4B and 4D). Single treatment with RN 1734 also increased the percentage of apoptotic cells in comparison to the control (Figure 4B and 4D). This finding appears to be consistent with the MTT cell proliferation assay where RN 1734 significantly decreased the proliferation of HT-29 cells (Figure 2B and 2C). However, the induction of apoptosis in this cell line was more pronounced with GSK1016790A than that of RN 1734 (Figure 4B and 4D). GSK1016790A-induced apoptosis in HT-29 cells was modestly attenuated by co-treatment with RN 1734, as illustrated by a slight decrease in the percentage of apoptotic cells at both time points tested (Figure 4B and 4D), further confirming the inhibitory action of RN 1734 on GSK1016790A-induced cell death. Altogether, these findings imply that both TRPV4 channel activation and inhibition could induce cell death in HT-29 cells, albeit at different degrees of cell death induction.
 |
Figure 4: Apoptosis assay using Annexin-V/PI double staining in HT-29 cells.
|
Representative images and quantitative analysis of apoptotic cells (expressed as percentage) at 24 hours (A and B) and 48 hours (C and D). Heat-induced cells were used as a positive control for the assay. Data are presented as mean ± standard deviation (n=3). Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple comparisons test, **P < 0.01, *** P < 0.001, **** P < 0.0001.
Discussion
The present in vitro study identified differential expression of TRPV4 in two human colorectal cancer cell lines, HT-29 and HCT-116 cells, compared with the non-transformed normal colon, CCD-18Co cells. Low expression of TRPV4 mRNA levels was observed in HT-29 cells while in HCT-116 cells, the TRPV4 mRNA level was undetectable by quantitative real-time PCR. A previous work by Sozucan et al.,21 has reported reduced expression of TRPV4 mRNA in the tissue samples derived from CRC patients compared with their normal tissues. The results obtained from the present study further extend this finding since TRPV4 was downregulated in HT-29 and HCT-116 cells in comparison to the normal colon cells. Indeed, altered expression of TRP channels has previously been described in a variety of cancer types,26, 27 whereby their expression levels could change in response to hormones, as exemplified by TRPV6 in T47D breast cancer cell lines28 or cancer stage-dependent as in the case of TRPM1 where its expression is higher in early stage melanomas but is lost as the disease progresses into a more aggressive phenotype.29 However, it remains to be determined if such changes in TRP expression levels are the primary steps in cancer progression or are secondary to other changes.27
Having shown that HT-29 cells expressed markedly low levels of TRPV4 mRNA, the potential role of remaining TRPV4 and its utility as a therapeutic target was explored by assessing the effect of TRPV4 pharmacological modulation on the proliferation of HT-29 cells. Previous studies have provided evidence to support a role for TRPV4 in regulating the proliferation of several types of cancer cells including the tumour endothelial cells30 and gastric cancer cells.31 The results of the present study showed that pharmacological activation of TRPV4 with a selective activator GSK1016790A augmented the proliferation of HT-29 cells. On the contrary, the proliferation of HT-29 cells was reduced in the presence of a TRPV4 inhibitor RN 1734. Co-treatment of GSK1016790A with RN 1734 appeared to hamper GSK1016790A-mediated increases in proliferation of HT-29 cells. The findings obtained from the present study are consistent with the results of a previous study by Ohashi and co-researchers32 who investigated the functional activity of TRPV4 in oligodendrocyte precursor cells (OPCs). The authors showed that TRPV4 is functionally expressed in OPCs, as evidenced by a sustained increase in intracellular Ca2+ upon stimulation with GSK1016790A. They also found that treatment with GSK1016790A promotes OPC proliferation in a concentration-dependent manner, which is inhibited by co-treatment with a TRPV4 antagonist HC067047. Further experiments uncovered that GSK1016790A-induced increases in proliferation of OPCs requires TRPV4-mediated Ca2+ influx and occurs via protein kinase C pathway.32
To determine the other functional roles of TRPV4 in colorectal cancer, we conducted a cell cycle analysis of DNA contents in HT-29 cells at 24 and 48 hours time points. No significant alteration in the cell cycle profile was observed when HT-29 cells were treated with GSK1016790A alone, RN 1734 alone or co-treated with GSK1016790A and RN 1734 in comparison to control. This observation implies that TRPV4 channels do not play a major role in cell cycle, at least in HT-29 cells. Our findings appear to be in line with previous work by Olivan-Viguera et al.,33 who reported that pharmacological activation of TRPV4 has no profound effects on the cell cycle progression of human melanoma A375 cells and human keratinocytes HaCaT cells, both of which functionally express TRPV4 channels.
Given reports on the role of TRPV4 activation-related cell death in several types of cancer cells,19, 33, 34 we then attempted to ascertain the functional consequences of TRPV4 pharmacological modulation on cell death in HT-29 cells. Our analysis from the Annexin-V/PI double-staining assay revealed that single treatment with GSK1016790A time-dependently induced cell death in HT-29 cells. Apoptotic cells have been shown to induce compensatory proliferation of neighbouring cells via the release of CrkI-containing microvesicles.35 This could explain why we observed increases in both proliferation and apoptosis in GSK1016790A-treated HT-29 cells. Similar induction of cell death was also observed in response to single treatment with the TRPV4 inhibitor RN 1734, which appears to be consistent with our observation in the proliferation of RN 1734-treated HT-29 cells. However, the induction of cell death as a consequence of TRPV4 inhibition in HT-29 cells was less pronounced than that of TRPV4 activation. Moreover, GSK1016790A-induced cell death was slightly decreased when RN 1734 was co-administered in HT-29 cells. The present data highlighted that pharmacological modulation of TRPV4 using either the activator or inhibitor could potentially induce cell death at varying degrees in HT-29 cells.
As far as the TRPV4 expression level is concerned, it is intriguing to note that, from our study, pharmacological activation of TRPV4 was capable of producing cell death in HT-29 cells with endogenously low TRPV4 levels. This is because TRPV4 activation has been shown to produce cell death in TRPV4 overexpressing basal breast cancer cells19 as well as in human melanoma A375 cells and human keratinocytes HaCaT cells with high endogenous levels of TRPV4,33, 34 consistent with the fact that high Ca2+ influx will lead to Ca2+ overload which eventually initiate cell death pathways.36 Considering that TRPV4 pharmacological activation, and to a lesser extent TRPV4 pharmacological inhibition, promoted cell death in HT-29 cells (which exhibited low endogenous levels of TRPV4), further studies are warranted to define the precise mechanism responsible for the induction of cell death in this cell line. Such studies would also be valuable in providing mechanistic insights into the downstream signalling pathways linking TRPV4 channels and cell death in colorectal cancer.
Conclusion
In conclusion, our work demonstrates that TRPV4 mRNA level is downregulated in HT-29 and HCT-116 colorectal cancer cells compared to normal colon cells. Moreover, modulation of TRPV4 channel by activation and inhibition using specific pharmacological modulators results in altered proliferation and varying degrees of induction of cell death in colorectal cancer cells with pronounced downregulation of TRPV4. Pharmacological targeting of TRPV4 may represent a new therapeutic approach for the treatment of CRC.
Acknowledgements
The authors would like to thank Prof. Gregory Monteith (The University of Queensland, Australia) for his valuable and constructive suggestions on the manuscript. The authors also thank Prof. Dr. Mohd Afandi Muhamad (Universiti Sultan Zainal Abidin) for his assistance in proofreading the manuscript, Prof. Dr. Abd Manaf Ali and Syed Ahmad Tajudin Tuan Johari (Faculty of Bioresources and Food Industry, Universiti Sultan Zainal Abidin) for their advice and technical assistance in using the flow cytometry for the cell cycle and cell death experiments and also Dr. Myat Moe Thwe Aung (Faculty of Medicine, Universiti Sultan Zainal Abidin) for her advice on statistical analyses. This work was supported by the Fundamental Research Grant Scheme (RR 202) from the Ministry of Education Malaysia (Higher Education).
Conflict of Interest
The authors declare no conflict of interest.
Funding Source
This work was funded by the Fundamental Research Grant Scheme (RR 202) from the Ministry of Education Malaysia (Higher Education).
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018 Nov;68(6):394-424.
- Grady WM, Markowitz SD. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig Dis Sci. 2015 Mar;60(3):762-72.
- House CD, Wang BD, Ceniccola K, Williams R, Simaan M, Olender J, et al. Voltage-gated Na+ Channel Activity Increases Colon Cancer Transcriptional Activity and Invasion Via Persistent MAPK Signaling. Sci Rep. 2015 Jun 22;5:11541.
- He T, Wang C, Zhang M, Zhang X, Zheng S, Linghu E, et al. Epigenetic regulation of voltage-gated potassium ion channel molecule Kv1.3 in mechanisms of colorectal cancer. Discov Med. 2017 Mar;23(126):155-62.
- Zhou ZH, Song JW, Li W, Liu X, Cao L, Wan LM, et al. The acid-sensing ion channel, ASIC2, promotes invasion and metastasis of colorectal cancer under acidosis by activating the calcineurin/NFAT1 axis. J Exp Clin Cancer Res. 2017 Sep 19;36(1):130.
- Lastraioli E, Iorio J, Arcangeli A. Ion channel expression as promising cancer biomarker. Biochim Biophys Acta. 2015;1848:2685-702.
- Zhang SS, Wen J, Yang F, Cai XL, Yang H, Luo KJ, et al. High expression of transient potential receptor C6 correlated with poor prognosis in patients with esophageal squamous cell carcinoma. Med Oncol. 2013;30(3):607.
- Nilius B. TRP channels in disease. Biochim Biophys Acta. 2007 Aug;1772(8):805-12.
- Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013 Feb;14(2):152-63.
- Holzer P. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacol Ther. 2011;131:142-70.
- Alessandri-Haber N, Dina OA, Yeh JJ, Parada CA, Reichling DB, Levine JD. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci. 2004;24:4444-52.
- Ye L, Kleiner S, Wu J, Sah R, Gupta RK, Banks AS, et al. TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell. 2012;151:96-110.
- Plant TD, Strotmann R. TRPV4: A Multifunctional Nonselective Cation Channel with Complex Regulation. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades: CRC Press; 2006. p. 139-54.
- Montell C. The TRP superfamily of cation channels. Sci STKE. 2005 Feb 22;2005(272):re3.
- Liedtke W, Tobin DM, Bargmann CI, Friedman JM. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2003 Nov 25;100 Suppl 2:14531-6.
- Chung MK, Lee H, Caterina MJ. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J Biol Chem. 2003 Aug 22;278(34):32037-46.
- Lee WH, Choong LY, Mon NN, Lu S, Lin Q, Pang B, et al. TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Sci Rep. 2016;6:27903.
- Lee WH, Choong LY, Jin TH, Mon NN, Chong S, Liew CS, et al. TRPV4 plays a role in breast cancer cell migration via Ca2+-dependent activation of AKT and downregulation of E-cadherin cell cortex protein. Oncogenesis. 2017 May 22;6(5):e338.
- Peters AA, Jamaludin SYN, Yapa KTDS, Chalmers S, Wiegmans AP, Lim HF, et al. Oncosis and apoptosis induction by activation of an overexpressed ion channel in breast cancer cells. Oncogene. 2017 Jul 31;36:6490-500.
- Wasilewski A, Krajewska U, Owczarek K, Lewandowska U, Fichna J. Fatty acid amide hydrolase (FAAH) inhibitor PF-3845 reduces viability, migration and invasiveness of human colon adenocarcinoma Colo-205 cell line: an in vitro study. Acta Biochim Pol. 2017;64(3):519-25.
- Sozucan Y, Kalender ME, Sari I, Suner A, Oztuzcu S, Arman K, et al. TRP genes family expression in colorectal cancer. Exp Oncol. 2015 Sep;37(3):208-12.
- Bahari NN, Jamaludin SYN, Jahidin AH, Zahary MN, Mohd Hilmi AB, Bakar NHA, et al. The emerging roles of TRPV4 in cancer. Biomed Pharmacol J. 2017;10:1757-64.
- Aung CS, Ye W, Plowman G, Peters AA, Monteith GR, Roberts-Thomson SJ. Plasma membrane calcium ATPase 4 and the remodeling of calcium homeostasis in human colon cancer cells. Carcinogenesis. 2009;30:1962-9.
- Ho K, Yazan LS, Ismail N, Ismail M. Apoptosis and cell cycle arrest of human colorectal cancer cell line HT-29 induced by vanillin. Cancer Epidemiol. 2009 Aug;33(2):155-60.
- Bundscherer A, Malsy M, Lange R, Hofmann P, Metterlein T, Graf BM, et al. Cell harvesting method influences results of apoptosis analysis by annexin V staining. Anticancer Res. 2013 Aug;33(8):3201-4.
- Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem. 2012;287:31666-73.
- Prevarskaya N, Zhang L, Barritt G. TRP channels in cancer. Biochim Biophys Acta. 2007 Aug;1772(8):937-46.
- Bolanz KA, Hediger MA, Landowski CP. The role of TRPV6 in breast carcinogenesis. Mol Cancer Ther. 2008 Feb;7(2):271-9.
- Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, Woolf EA, et al. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 1998 Apr 1;58(7):1515-20.
- Thoppil RJ, Adapala RK, Cappelli HC, Kondeti V, Dudley AC, Gary Meszaros J, et al. TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Sci Rep. 2015 Sep 21;5:14257.
- Xie R, Xu J, Xiao YF, Wu J, Wan H, Tang B, et al. Calcium promotes human gastric cancer via a novel coupling of calcium-sensing receptor and TRPV4 channel. Cancer Res. 2017 Sep 26.
- Ohashi K, Deyashiki A, Miyake T, Nagayasu K, Shibasaki K, Shirakawa H, et al. TRPV4 is functionally expressed in oligodendrocyte precursor cells and increases their proliferation. Pflugers Arch. 2018;470:705-16.
- Olivan-Viguera A, Garcia-Otin AL, Lozano-Gerona J, Abarca-Lachen E, Garcia-Malinis AJ, Hamilton KL, et al. Pharmacological activation of TRPV4 produces immediate cell damage and induction of apoptosis in human melanoma cells and HaCaT keratinocytes. PLoS One. 2018;13(1):e0190307.
- Zheng J, Liu F, Du S, Li M, Wu T, Tan X, et al. Mechanism for Regulation of Melanoma Cell Death via Activation of Thermo-TRPV4 and TRPV2. J Oncol. 2019;2019:7362875.
- Gupta KH, Goldufsky JW, Wood SJ, Tardi NJ, Moorthy GS, Gilbert DZ, et al. Apoptosis and Compensatory Proliferation Signaling Are Coupled by CrkI-Containing Microvesicles. Dev Cell. 2017 Jun 19;41(6):674-84 e5.
- Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008 Oct 27;27(50):6407-18.

