Identification of Paulownin as a Potential Monoamine Oxidase-B (MAO-B) Inhibitor by Screening the Traditional Chinese Medicine (TCM) Database
 , Suhad Sami Humadi2, and Azher Abdulmutaleb Ibrahim1
, Suhad Sami Humadi2, and Azher Abdulmutaleb Ibrahim1 1Department of Pharmacology, College of pharmacy, Al-Zahrawi University, Karbala, Iraq.
2Department of Pharmacognosy, College of pharmacy, Al-Zahrawi University, Karbala, Iraq.
Corresponding Author E-mail:hodhar3@gmail.com
DOI : http://dx.doi.org/10.13005/bpj/3447
Download this article as:
![]()
One of the prominent neurodegenerative ailments that can inflict the elderly population is the Parkinson’s disease (PD). This disease is marked by the reduction of dopamine concentration inside brain due to the gradual destruction of the dopaminergic neurons. The treatment of PD is primarily related to symptoms mitigation, yet no treatment is available to restrain or stop the development of neuronal degeneration. In this regard, it is believed that the selective block of monoamine oxidase type B (MAO-B) enzyme can’t only reduce the metabolism of neuronal dopamine but also lower the production of the side product hydrogen peroxide. Thus, the inhibition of MAO-B enzyme may have a neuroprotective influence by limiting oxidative stress related to the release of hydrogen peroxide. Therefore, the goal of this in-silico research is to specify a selective MAO-B inhibitor by screening the Traditional Chinese Medicine (TCM) database through application of docking as well as molecular dynamics (MD) tools. As a result, it is expected that the herbal compound Paulownin may have the capacity to selectively inhibit MAO-B enzyme. In this regard, the MD simulation indicates that Paulownin has a close proximity to MAO-B active site with a high binding energy as compared to that of MAO-A. Additionally, docking image examination refers to the fact that this compound may be involved in binding with important residues of MAO-B active site. Further, prediction of several chemical features indicates that the compound may have good oral absorption. Also, Paulownin is expected to be water soluble, non-mutagenic, safe and it may have a moderate volume of distribution and CNS permeability inside the body. Lastly, the compound Paulowninwas able to record an IC50 (half maximal inhibitory concentration) of 42.6 µM when tested against MAO-B enzyme in the monoamine oxidase inhibition assay.
KEYWORDS:Docking; Dynamics simulation; Paulownin; MAO-B; TCM
Introduction
Parkinson’s disease (PD) is a multisystem and a gradual disorder where the dopaminergic neurons in the midbrain region are specifically degenerated.1 As the incidence of PD is growing with age, this neurodegenerative disease is mainly affecting elderly population.2 Clinically, PD diagnosis depends on the manifestation of fundamental motor symptoms and these are: rigidity of muscles, posture imbalance, bradykinesia and body tremor. Also, non-motor symptoms like cognition and sleep disorders may develop during the disease course. And sometimes these non-motor signs may predate the incidence of PD motor features by several years.3 In addition to the degeneration of dopaminergic nerve cells, PD is also marked by the accumulation of misfolded proteins inside neurons. These neuronal protein inclusions are known as Lewy bodies that composed of α-synuclein protein.4 Pathologically, the buildup of α-synuclein can disrupt normal functions of the neurons and eventually lead to progressive degeneration.5 It is thought that this neuronal degeneration can precede the onset of PD clinical symptoms by several decades.6 The diagnosis of PD is mainly dependent on the clinical characteristics of the disease in addition to the employment of specialized brain imaging tools.7 The exact reason for PD neurodegeneration is not defined yet, and the underlying pathology framework appears to be complicated with the interplay of multiple risk factors.8 Mutations in specific genes like those encoding for α-synuclein (SNCA) or leucine rich repeat kinase-2 (LRRK2) can predispose to the incidence of familial PD.9 Moreover, the exposure to head trauma or other environmental agents like pesticides can increase the likelihood of idiopathic PD.10 While the available therapeutic options like the stimulation of deep brain or the use of dopaminergic agents can only alleviate PD motor symptoms, no treatment is still available to pause or slow the progress of PD degeneration.11,12 It is assumed that no protective agent was yet identified because all the related clinical trials were focused on volunteers diagnosed with PD at the advent of motor symptoms. It is well-known that the PD motor symptoms usually emerge when about 75% of the dopaminergic neurons are already destructed in the substantia nigra region of the brain. So, the neuronal degeneration will be at an advanced level when PD is clinically diagnosed and as such the effect of protective agents will be limited at that point.1 As the PD pathology is complex, then the presentation of a drug candidate that can target more than one molecular target or pathway seems to be an interesting strategy. In this perspective, it is believed that the selective block of the monoamine oxidase-B (MAO-B) enzyme can have a dual effect on PD pathology by limiting the oxidative deamination process of both endogenous and exogenous dopamine while reducing the oxidative stress caused by the hydrogen peroxide byproduct. Therefore, the administration of selective MAO-B inhibitors is expected to both lower the daily dose of L-dopa and offer some level of neuroprotection in PD patients.13 Due to this dual beneficial effect of MAO-B inhibition, this computational study was designed to screen the Traditional Chinese Medicine (TCM) online compounds for their probable capacity to selectively block the MAO-B enzyme. The current in-silico study has utilized several tools that encompass molecular docking, dynamics simulation and in-vitro enzyme inhibition assay to evaluate the MAO-B inhibition potential for these TCM compounds.
Material and methods
Planning of virtual screening paradigm
The computerized screening of TCM compounds for their MAO-B inhibition potential in this study is made up of four major steps: molecular docking of the TCM phytocompounds against MAO-B enzyme, then anticipation of pharmacokinetics, toxicity and chemical characteristics for only the best five hits, followed by a 50 nanoseconds (ns) dynamics simulation study for these five compounds, with a determination of the MAO-B median inhibitory concentration (IC50) for the best hit as a final step by using an in-vitro enzyme inhibition assay. Additionally, these selected top hits were subjected to a molecular docking and dynamics simulation against MAO-A enzyme. The generated docking and dynamics simulation results for these hit compounds were compared between MAO-A and MAO-B enzymes to ensure the potential selectivity of these compounds. As a summary, the applied methodology in this screening is very identical to what was implemented in our formerly published studies.14,15
Molecular docking study
At start, the TCM database of herbal compounds were screened against MAO-B active site by using the DrugRep online server.16 For this step, only chain A of the human monoamine oxidase B crystal (PDB: 2BK3) was submitted to the virtual screening server.17 Then, the docking coordinates were specified as X: 56, Y: 153 and Z: 23 while measurements of the grid box were 22*22*22 Angstrom (Å). It is worth mentioning that these applied coordinates and grid box dimensions are the same to what had been used in previously published studies.18,19 After that, the docking of 2,390 compounds in the TCM library was performed by the DrugRep server which utilize both the AutoDockTools (ADT) 1.5.6 and AutoDock Vina 1.1.2 for this purpose.20,21 Finally, the output hits were tabulated and ordered based on their computed energy of binding to target enzyme. And as such, only the best five TCM compounds with the smallest energy were picked for more computerized assessment. Furthermore, the selectivity of these best hits for MAO-B inhibition was considered by also docking of these compounds against MAO-A enzyme (PDB: 2BXR) using docking measurements of X: 21, Y: 2 and Z: 6.22 Then, the predicted affinity of these compounds towards MAO-A and MAO-B enzymes were compared in terms of docking energy and orientation of docking pose. And for each docking complex, the orientation of the compound pose with the lowest energy of binding was observed by using discovery studio visualizer 21.1.0 and PyMOL 2.4.1. It is worth mentioning that the precision of this docking step was calculated for both MAO-A and MAO-B targets by applying the redocking procedure. In this process, the co-crystalized ligand was first taken out of target enzyme then docked again by using the same virtual conditions. After that, the conformational changes between co-crystalized and docked compound was computed as the root mean square deviation (RMSD) value.
Prediction of the pharmacokinetics, chemical and toxicity qualities
For this stage of the virtual study, both pkCSM and SwissADME websites were used to expect the chemical and pharmacokinetics features for the best hit compounds.23,24 Also, the ProTox 3.0 online tool was employed to predict each hit toxicity potential while the Molsoft L.L.C. site was utilized to compute drug-likeness score.25 In addition, the herbal origin and documented pharmacological effect were reviewed for these selected hits.
Study of molecular dynamics (MD) simulation
It is widely known that molecular docking only offers a static or a frozen representation for the interaction between ligand under assessment and target protein. And to obtain a dynamic view for the ligand binding, the molecular dynamics (MD) study usually applies multiple forcefields to compute and track molecules and atoms movement during simulation time. Consequently, the MD study was utilized in this in-silico screening to get a better understanding for the interaction between each ligand and target.26 For this objective, the YASARA Dynamics 20.12.24 software was used to do the simulation study.27 In this stage, the docking complex between each hit and MAO-B with the lowest energy pose was subjected for 50 ns simulation. The applied simulation parameters and methodology are identical to what was used in previously published manuscripts.14,15 Once more, the MD simulation of each hit was also evaluated against MAO-A throughout 50 ns duration and results were compared with that of MAO-B. The aim of this comparison is to estimate each compound selectivity to inhibit MAO-B enzyme. In addition, the docked Farnesol and Clorgyline were included as a control for the simulation of MAO-B and MAO-A respectively. Then, the final simulation results were presented as mean RMSD value of ligand movement in relation to enzyme active site, while the binding energy was calculated as average molecular mechanic Poisson Boltzmann surface area (MM-PBSA).
Monoamine oxidase inhibition assay
In this in-vitro assay, the IC50 value was measured for the best hit as an indicator for the compound’s inhibition potential against MAO-B. The basic principle of the monoamine oxidase inhibition assay depends on the ability of the enzyme to metabolize the non-fluorescent kynuramine into the fluorescent 4-hydroxyquinoline. As such, any inhibitor compound can interfere with this metabolic pathway and reduce the concentration of the fluorescent product.28 In this assay, the compound under investigation was purchased from Cayman chemical online store andserially dilutedwith dimethyl sulfoxide (DMSO) through eight points of a 96-well plate. Then, an assay kit was purchased from Sigma-Aldrich (MAK136) and utilized to carry out the monoamine oxidase inhibition test.In this assay, MAO-B enzyme was added to these eight points in a concentration of 0.015 mg/ml with the presence of 20 μM of the non-fluorescent kynuramine.This mixture was incubated for twenty minutes at a temperature of 37 °C then the fluorescence activity of the 4-hydroxyquinoline was recorded by a fluorometer at a wavelength pair of 310/ 380 nm. Additionally, the well-known antiparkinsonian agent Zonisamide was included in this assay as a positive control. The measurement of each point in the dose-inhibition curve was repeated as a duplicate and plotted as mean ± standard deviation.
Results
Before screening compounds database in the TCM library, the redocking approach was utilized to ensure the fitness of the applied docking procedure. In this validation approach, the co-crystalized Farnesol was taken off the MAO-B active site. Then, the removed Farnesol was docked again into MAO-B using the same docking protocol employed in the screening of TCM library. And by aligning the co-crystalized and docked Farnesol, the precision of the docking protocol can be concluded as RMSD value of the conformational variations. As noted in Figure 1 (A), the computed RMSD of conformational changes was only 0.8 Å when comparing the co-crystalized Farnesol versus the docked one. Also, the energy of binding was recorded to be -7.96 Kcal/ mol for the docking of Farnesol against MAO-B. As mentioned before, the MAO-A enzyme was used as an additional target during docking and simulation to assess the selectivity of the best screening hits against MAO-B. As such, the redocking method was used also to evaluate the accuracy of the employed protocol against MAO-A. According to Figure 1 (B), the conformational changes RMSD was 0.2 Å for the docked and co-crystalized Clorgyline while the docking energy was reported to be -6.67 Kcal/ mol.
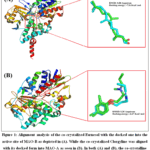 |
Figure 1: Alignment analysis of the co-crystalized Farnesol with the docked one into the active site of MAO-B as depicted in (A). While the co-crystalized Clorgyline was aligned with its docked form into MAO-A as seen in (B). In both (A) and (B), the co-crystalline ligand is shown in cyan color while the docked one is colored by green. |
After conducting the virtual screening of the TCM compounds against MAO-B, the generated hit compounds were listed based on the calculated energy of binding. In this computational study, as seen in Table 1, only the best five herbal compounds against MAO-B with the least docking energy were presented. In this table, the docking energy of these best five compounds was also computed toward MAO-A to consider ligand selectivity against target enzyme. As noted from Table 1, the docking energy for these five hits seem to be slightly higher for MAO-B as compared to MAO-A. Moreover, all the listed docking energy values in Table 1 appear to be more than 10 Kcal/ mol regardless of enzyme subtype.
Table 1: Comparison of docking energy for the best screening compounds against MAO-A and MAO-B monomers.
| No. | Hit ID | Docking energy (Kcal/ mol) | |
| MAO-A | MAO-B | ||
| 1 | Licoflavone B | -11.4 | -12.7 |
| 2 | Corylin | -10.9 | -11.9 |
| 3 | Oxysanguinarine | -10.9 | -11.7 |
| 4 | Paulownin | -11.0 | -11.5 |
| 5 | Rutaecarpine | -10.7 | -11.3 |
Then, the best screening hits against MAO-B were reviewed for their existing herbal sources and the documented pharmacological activities as listed in Table 2. It is obvious from this table that these listed hits have anticancer, anti-inflammatory or antimicrobial actions. And with the exception of the lignan Paulownin, these hits are either flavonoid or alkaloid medicinal compounds.
Table 2: A tabular summary of the herbal origin and the probable pharmacological effects for the best hits acquired by the structure-based computational screening.
| No. | Phytomedicine name | Herbal origin | Pharmacological effect |
| 1 | Licoflavone B | Glycyrrhiza inflata | Anti-inflammatory,29 anticancer,30 anti-schistosomiasis 31 |
| 2 | Corylin | Psoralea corylifolia | Osteogenic,32 anticancer,33 anti-hyperlipidemia 34 |
| 3 | Oxysanguinarine | Macleaya cordata, Meconopsissimplicifolia, Sanguinaria canadensis | Anti-inflammatory,35 anticancer,36 antiviral,37 antimalarial 38 |
| 4 | Paulownin | Paulownia tomentosa | Anticancer,39 antimicrobial 40 |
| 5 | Rutaecarpine | Evodia rutaecarpa | Cardiotonic,41 anti-inflammatory,42 antioxidant 43 |
A predictive summary for the important chemical properties that can affect the oral bioavailability of the five hit compounds is presented in Table 3. According to this table, it is very easy to recognize that all the hit compounds but Licoflavone B do have chemical features that align with the rule of five (RO5) conditions.44 And when considering the RO5 cutoff limits, then it is clear that all these natural compounds do have a molecular weight less than 500 g/ mol. Also, all these compounds are expected to have below than 5 H-bond donor groups and also lower than 10 H-bond acceptor groups. In addition, all the listed hits with the exception of Licoflavone B seem to have an octanol/ water partition coefficient of no more than 5. Besides, all the compounds in Table 3 are predicted to have a polar surface area (PSA) of less than 120 Å2 and also less than 10 rotatable bonds in its structure. Finally, the listed molecular formula column reveals that both Licoflavone B and Corylin are flavonoids while Oxysanguinarine and Rutaecarpine are alkaloids. On the other hand, the fourth hit Paulownin appears to be a lignan compound.
Table 3: A tabular presentation of the chemical properties for the best phytomedicines acquired by the computerized screening of the Monoamine Oxidase B (MAO-B) with the Traditional Chinese Medicine (TCM) database. These TCM compounds were listed based on the lowest docking energy.
| No. | Hit name | Molecular formula | M.W. (g/mol) | HBD | HBA | PSA (Å2) | Log P | Rotatable bonds no. |
| 1 | Licoflavone B | C25H26O4 | 390.5 | 2 | 4 | 70.67 | 5.19 | 5 |
| 2 | Corylin | C20H16O4 | 320.3 | 1 | 4 | 59.67 | 3.53 | 1 |
| 3 | Oxysanguinarine | C20H13NO5 | 347.3 | 0 | 5 | 58.92 | 3.31 | 0 |
| 4 | Paulownin | C20H18O7 | 370.4 | 1 | 7 | 75.61 | 2.10 | 2 |
| 5 | Rutaecarpine | C18H13N3O | 287.3 | 1 | 2 | 50.68 | 3.02 | 0 |
M.W.: the molecular weight; PSA: thepolar surface area; HBA: theH-bond acceptor group; HBD: theH-bond donor group;Å: degree angstrom; Log P: the logarithmic value of partition coefficient.
After that, the drug-likeness numerical score was calculated for each of the five hits as seen in Table 4. As shown in this table, all of the hit compounds with the exception of Licoflavone B are anticipated to have physicochemical features that not resemble those of known drug molecules as was inferred by their low drug-likeness scores. Regarding the pharmacokinetics profile, it is predicted that all these hits are highly or moderately soluble in water with the exception of Licoflavone B. Moreover, all the listed hits are believed to have a high gastrointestinal absorption however the expected volume of distribution inside body was moderate for compounds Oxysanguinarine and Paulownin. Also, the prediction of the ratio for the brain/ blood concentration points to the possibility that all listed compounds in Table 4 are CNS permeable with Log BB value of more than -1. Finally, the prediction of toxicity potential points out to the likelihood that these herbal compounds are slightly toxic with an LD50 value of 1000 mg/ Kg or more. Nonetheless, the listed hits but Paulownin are anticipated to have a mutagenic ability as indicated by Ames toxicity column in Table 4. In this direction, it was predicted that all these hits except Paulownin may have the ability to block multiple variant forms of cytochrome P450.
Table 4: A prediction summary of the pharmacokinetics, toxicity characteristics and drug-likeness score for the best screening hits. These compounds were organized in this table according to the lowest energy of binding as computed by docking process.
| No. | hit ID | Drug-likeness | Pharmacokinetics | Toxicity | ||||
| Solubility in water (mg/ml) | % of GI absorption | VDss(L/Kg) | Log BB | Ames toxicity | LD50(mg/ Kg) | |||
| 1 | Licoflavone B | 0.51 | 9.96e-06 (Poor) | 92.15 | 1.17 | 0.21 | Yes | 2,570 |
| 2 | Corylin | 0.15 | 6.46e-03 (Moderate) | 96.86 | 2.69 | 0.18 | Yes | 3,850 |
| 3 | Oxysanguinarine | -1.35 | 1.26e-02 (Moderate) | 100.00 | 0.54 | -0.46 | Yes | 1,000 |
| 4 | Paulownin | -0.65 | 5.60e-01 (Soluble) | 97.12 | 0.55 | -0.57 | No | 1,500 |
| 5 | Rutaecarpine | 0.05 | 5.00e-02 (Soluble) | 97.29 | 0.87 | 0.67 | Yes | 1,400 |
GI: gastrointestinal;VDss: volume of distribution in the steady state; Log BB: logarithm of brain to blood concentration ratio; LD50: median value of lethal dose.
And to get a better understanding for the ligand-target interaction beyond the scope of docking study, the MD process was done to assess the evolution of each ligand-enzyme complex over 50 ns duration. A tabular summary for the output of the MD simulation is reported in Table 5. As observed in this table, for each hit, the mean ligand movement and the average binding energy were compared between MAO-A and MAO-B isoenzymes to assess the selectivity of each compound. Based on Table 5, it is obvious to note that all the hits have a closer proximity to MAO-B active site as compared to that of MAO-A. Also, the average binding energy computed during simulation was higher for the binding of these hits to MAO-B enzyme. But the highest MAO-B binding selectivity was recorded to the compound Paulownin with more than 2.2- and 2.9-folds of MAO-B preference when considering ligand proximity and MM-PBSA binding energy respectively. As indicated in Table 5, both Farnesol and Clorgyline were included as positive controls for MAO-B and MAO-A simulations. And it is worth to notice that the MM-PBSA binding energy for both Farnesol and Clorgyline were higher than that computed for the listed five compounds.
Table 5: A summary analysis of the MD simulation study for the best TCM phytocompounds acquired by the virtual screening.
| No. | Phytomedicine name | RMSD for mean ligand movement (Å) | Average value for MM-PBSA binding energy (Kcal/ mol) | ||
| MAO-B | MAO-A | MAO-B | MAO-A | ||
| 1 | Licoflavone B | 1.36 | 2.31 | 19.91 | 17.16 |
| 2 | Corylin | 1.76 | 2.93 | 13.30 | 12.11 |
| 3 | Oxysanguinarine | 2.43 | 3.79 | 13.48 | 2.06 |
| 4 | Paulownin | 1.52 | 3.39 | 18.53 | 6.27 |
| 5 | Rutaecarpine | 2.46 | 3.87 | 11.91 | 4.82 |
| 6 | Farnesol | 3.10 | – | 28.69 | – |
| 7 | Clorgyline | – | 2.50 | – | 24.00 |
MD: themolecular dynamics; Å: angstrom; RMSD: the root mean squared deviation; MM-PBSA: themolecular mechanic Poisson Boltzmann surface area.
In order to obtain a deep understanding for the attitude of each compound during its simulation, stepwise plots for the ligands’ movement RMSD throughout 50 ns duration were presented in Figure 2 for MAO-B and MAO-A. According to Figure 2, it is very clear to say that the compound Paulownin did record a remarkable MAO-B binding selectivity of more than 2 times as compared to MAO-A especially after 15 ns simulation duration. On the contrary, the compoundsLicoflavone B and Corylin have shown a lower binding preference to MAO-B enzyme when considering the average ligand movement RMSD in Figure 2. Moreover, both Oxysanguinarine andRutaecarpinedid present the least preference to MAO-B binding with significant fluctuations in ligand proximity to enzyme active site during MD simulation.
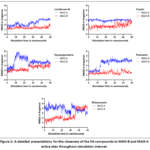 |
Figure 2: A detailed presentations for the closeness of the hit compounds to MAO-B and MAO-A active sites throughout simulation interval. |
Then, the most selective hit (Paulownin) was validated in-vitro for its ability to inhibit MAO-B enzyme. According to the dose-inhibition curve in Figure 3, the calculated IC50 for Paulownin was 42.6 µM. This inhibition capacity of Paulownin seemed to be less than that of Zonisamide which was able to achieve an IC50 of 28.9 µM in the MAO inhibition assay. Moreover, the R2 (goodness of fit) for the nonlinear curve fitting in Figure 3 were 94% and 99% for Paulownin and Zonisamide respectively.
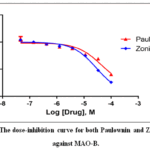 |
Figure 3: The dose-inhibition curve for both Paulownin and Zonisamide against MAO-B. |
As a result for the prediction and simulation analyses, the compound Paulownin appears to have a non-mutagenic nature and a high MAO-B binding selectivity. As such, Paulownin was selected for an in-depth evaluation of its docking complex as illustrated in Figure 4. Interestingly, according to this figure, the compound Paulownin seems to be engaged in specific interactions with essential MAO-B active site residues like Ile 199, Tyr 326, Tyr 398 and Tyr 435.
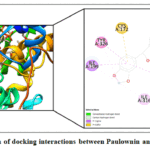 |
Figure 4: Visualization of docking interactions between Paulownin and MAO-B active site. |
Discussion
Generally, the management of PD is primarily focused on the alleviation of motor manifestations through administration of L-dopa to replace the dopamine level inside CNS. However, the replacement of the dopamine can be linked with motor complications like dyskinesia. One of the pharmacotherapeutic options to overcome these motor complications is to use a selective MAO-B inhibitor as an additive agent. Adding an inhibitor of MAO-B to PD treatment regimen can extend the duration of the administered L-dopa dose and also boost its effect.45 MAO enzyme is present in two isoform types, MAO-A and MAO-B, that utilize a flavin adenine dinucleotide (FAD) moiety as a cofactor to participate in dopamine metabolism into the corresponding aldehyde product.46 During this oxidative deamination pathway, the hydrogen peroxide (H2O2) is formed as a side product. The formation of H2O2 can participate in the cellular oxidative stress. Unlike MAO-A, the activity of MAO-B enzyme is usually upregulated with age advances and progress of neurodegeneration. As a result, it was hypothesized that the selective inhibition of only MAO-B enzyme in PD patients can elevate the CNS concentration of dopamine and also lower the production of H2O2.47 Consequently, in the current study, a library of herbal compounds in the TCM database was screened against MAO-B enzyme. Both docking and MD simulation programs were utilized in this computational project to recognize a potential MAO-B inhibitor. Moreover, a machine learning based tool was utilized to predict MAO-B IC50 values for the best screening hits.
At first, the accuracy of the applied virtual screening methodology and parameters were evaluated by using the redocking approach. Based on the redocking outputs in Figure 1, the conformational change RMSD was only 0.8 Å between the naive and docked Farnesol when considering the MAO-B crystal. Also, a low RMSD for the variation in conformation of 0.2 Å was computed by aligning the docked and co-crystalized Clorgyline in the case of MAO-A target. As such, it is predicted that the applied docking approach in this study is accurate and reliable due to the computed low conformational change RMSD.48
After finishing the virtual screening of the compounds in TCM database against MAO-B, the output hits were organized according to their energy of binding. As indicated in Table 1, only the best five hits with the least energy of binding were listed. And to consider the selectivity of each hit, the MAO-A docking energy was also included in Table 1. It can be noticed from this table that the energy of binding is slightly higher for MAO-B enzyme as compared to MAO-A. Therefore, these findings in Table 1 can refer to the possible slight selectivity of these five hits against MAO-B. However, further verification may be necessary by using MD simulation to assess the selectivity of these selected hits.
Then, these five hit compounds were reviewed in Table 2 for their natural source and documented biological effects. Dependingon this table, these listed hits are supposed to possess anticancer, anti-inflammatory or antimicrobial activities. Additionally, several chemical characteristics were anticipated for these five hits as seen in Table 3. And by considering the chemical formula in Table 3, then it can be observed that these compounds are either flavonoids or alkaloids with the exclusion of the lignan Paulownin. Also, it is predicted that most of these hits do have a good oral absorption due to the alignment of their chemical features in Table 3 with the Lipinski’s rule of five (RO5) conditions.44 The single violation for RO5 conditions can be noted for the first hit Licoflavone B, which has a Log P value of more than 5. In the same direction, it is believed that these listed compounds do have a reduced molecular flexibility with less than 10 rotatable bonds. Moreover, the topological polar surface area (PSA) for these compounds in Table 3 is expected to be lower than 120 Å2. Consequently, the reduced molecular flexibility and polar surface area can suggest a good oral bioavailability for these candidates.49
In the next stage of this in-silico study, the drug-likeness degree was calculated to each hit as shown in Table 4. Based on this table, all the listed hits except Licoflavone B were predicted to have a low drug-likeness degree. This finding refers to the fact that these compounds may have chemical and physical properties that are not similar to that of the already approved drugs. Also, in Table 4, the pharmacokinetics profile for these hits was evaluated by predicting the water solubility, intestinal absorption and volume of distribution inside body. In this regard, it is expected that the listed compounds are hydrophilic with a good or moderate water solubility. The only exclusion is the first compound Licoflavone B, which is anticipated to has a poor water solubility. This falls in agreement with the predicted Log P value for the Licoflavone B that was slightly higher than 5 as mentioned before in Table 3. Also, all the presented hits in Table 4 are believed to have a high degree of gastrointestinal absorption but the steady state volume of distribution (VDss) was moderate for both Oxysanguinarine and Paulownin. It is well-known that compounds with a moderate VDss can distribute equally through body tissues and blood.50 In addition, it is expected that these best five hits are CNS permeable as the theoretical log BB values for these compounds were higher than -1. It is believed that a log BB value of more than -1 can refers to a moderate or high permeability of the tested compounds according to previously established BBB permeability models.51 Finally, the assessment of toxicity potential in Table 4 indicated that these hits may be slightly toxic with an LD50 value of 1000 mg/ Kg or more.52 On the contrast, all the listed hits but Paulownin are anticipated to be mutagenic and have the capacity to interact and inhibit multiple variants of CYP450 enzyme.
And to gain a dynamic understanding for the evolution of each hit-target complex, the MD simulation stage was performed with the aid of multiple forcefields for 50 ns duration. In this study, both mean hit movement and average simulation binding energy were reported in Table 5 for both MAO-A and MAO-B enzymes. When considering the mean hit movement RMSD value in Table 5, it is obvious that this parameter was lower for MAO-B as compared to MAO-A. This can point to the possibility of closer hit nearness to MAO-B active site, consequently a stronger binding can be inferred in this case. In addition, a higher average MM-PBSA binding energy was calculated for the interaction of each hit with MAO-B enzyme. As such, it is expected that these five hits may have a higher preference toward MAO-B enzyme as indicated by the closer hit proximity to enzyme active site and the higher binding energy. However, the binding of these five hits to MAO-B may be weaker than Farnesol as the computed average binding energy for this control compound in Table 5 was higher. Interestingly, it seems that the compound Paulownin is the most MAO-B selective candidate. Depending on Table 5, this compound is anticipated to has more than 2.2 and 2.9 times of MAO-B selectivity when considering the ligand closeness to enzyme active site and the simulation binding energy respectively. This summary analysis of MD study in Table 5 can be more confirmed by inspecting the detailed plot of the ligand motion as a function of the simulation time duration as seen in Figure 2 for MAO-B and MAO-A.
It is well-known that the application of molecular docking is limited by multiple constraints like inaccurate scoring algorithm and possible flaws in predicting the solvation and target flexibility effects.53 Moreover, the dynamics simulation tools are known to rely on empirical force fields to estimate atomic interactions and this may lead to some inaccuracy in the calculations of binding affinity.54 Also, the use of predictive models may lead to flawed outputs in some instances due to overfitting as these models were built on limited experimental datasets with a confined application area. As such, the current theoretical findings must be considered as preliminary findings to be further validated by appropriate in-vitro laboratory testing.
Consequently, the hit compound Paulownin was more validated inside laboratory by the monoamine oxidase inhibition assay. In this in-vitro assay, different titrations of the compound were evaluated for its capacity to inhibit MAO-B activity. Zonisamide, a well-known antiepileptic and antiparkinsonian medication, was included as a positive control in this in-vitro assay. Based on the results of this assay, the inhibition capacity of Paulownin against MAO-B was inferior when compared to Zonisamide with a computed IC50 value of 42.6 and 28.9 µM respectively.
Furthermore, a careful examination of the docking complex for the most MAO-B selective candidate revealed that the ligand Paulownin may be involved in interactions with several essential residues in MAO-B active site. As noted by Figure 4, the compound Paulownin is engaged in hydrophobic interactions with the gating residues Ile 199, Tyr 326 and the aromatic cage residues Tyr 398, Tyr 435.55,56 These interactions suggest that the compound Paulownin may occupy a location within MAO-B active site that ensure enzyme inhibition.
Conclusion
MAO-B enzyme inhibitors are usually used as additive options in Parkinson’s disease (PD) treatment to reduce the daily dose of L-dopa therapy and thereby minimizing the associated motor complications. It is believed that the inhibition of MAO-B can decrease the metabolism of CNS dopamine while lowering the release of H2O2 as a side product. Thus, the selective block of MAO-B enzyme can have a neuroprotective effect by hindering the production of the oxidizing agent H2O2. Therefore, in this in-silico study, both dynamics simulation and docking were utilized to recognize a selective inhibitor of MAO-B enzyme among phytocompounds in the Traditional Chinese Medicine (TCM) database. As a result, it is believed that the lignan compound Paulownin may be the most selective inhibitor of MAO-B. The compound appears to have a close proximity to MAO-B active site with a high binding energy during simulation analysis. Besides, the compound Paulownin is believed to be implicated in several interactions with MAO-B active site key residues as observed by the docking study. Moreover, the prediction of chemical features revealed that Paulownin may have good oral absorption as it not violating any condition in the rule of five (RO5). Also, this hit is supposed to be water soluble, safe, non-mutagenic and it may have a moderate volume of distribution and CNS permeability. Lastly, the in-vitro monoamine oxidase inhibition assay had shown that the compound Paulowninwas able to record an IC50 value of 42.6 µM against MAO-B.
Acknowledgment
The author(s) would like to thanks the college of pharmacy, Al-Zahrawi University for their support of this work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable
Author Contributions
- Hasanain Abdulhameed Odhar: Conceptualization, Methodology, Data collection, Analysis, Writing – Original Draft.
- Suhad Sami Humadi: Writing – Review & Editing, Supervision.
- AzherAbdulmutaleb Ibrahim: Methodology, Review & Editing.
References
- Tanner CM, Ostrem JL. Parkinson’s Disease. Ropper AH, ed. New England Journal of Medicine. 2024;391(5):442-452.
CrossRef - Radhakrishnan DM, Goyal V. Parkinson’s disease: A review. Neurol India. 2018;66(7):S26-S35.
CrossRef - Sveinbjornsdottir S. The clinical symptoms of Parkinson’s disease. J Neurochem. 2016;139 Suppl 1:318-324.
CrossRef - Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8(12):1150-1157.
CrossRef - Power JHT, Barnes OL, Chegini F. Lewy Bodies and the Mechanisms of Neuronal Cell Death in Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathology. 2016;27(1):3.
CrossRef - Miller DB, O’Callaghan JP. Biomarkers of Parkinson’s disease: Present and future. Metabolism. 2015;64(3):S40-S46.
CrossRef - Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30(12):1591-1601.
CrossRef - Kalia L V., Lang AE. Parkinson’s disease. The Lancet. 2015;386(9996):896-912.
CrossRef - Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev. 2011;91(4):1161-1218.
CrossRef - Grotewold N, Albin RL. Update: Protective and risk factors for Parkinson disease. Parkinsonism RelatDisord. 2024;125.
CrossRef - Wójcik R, Dębska A, Zaczkowski K, et al. Deep Brain Stimulation for Parkinson’s Disease—A Narrative Review. Biomedicines 2025, Vol 13,. 2025;13(10).
CrossRef - Pringsheim T, Day GS, Smith DB, et al. Dopaminergic Therapy for Motor Symptoms in Early Parkinson Disease Practice Guideline Summary: A Report of the AAN Guideline Subcommittee. Neurology. 2021;97(20):942.
CrossRef - Chandra Sekar PK, Thomas SM, Veerabathiran R. An overview of the role of monoamine oxidase-B in Parkinson’s disease: implications for neurodegeneration and therapy. Open Exploration 2019 4:4. 2024;4(4):308-318.
CrossRef - Odhar HA. Screening of Traditional Chinese Medicine (TCM) Compounds for their Aromatase Inhibitory Potential by Applying Molecular Docking, Dynamics Simulation and Cytotoxicity Assay. Biomedical and Pharmacology Journal. 2025;18(3):2202-2213.
CrossRef - Odhar HA, Odhar ZA, Muhi MR. Screening of Traditional Chinese Medicine Library Against Penicillin-binding Protein 2a for Methicillin-resistant Staphylococcus aureus by Molecular Docking, Dynamics Simulation and In vitro Antimicrobial Activity. Biomedical and Pharmacology Journal. 2025;18(1):823-834.
CrossRef - Gan J hong, Liu J xiang, Liu Y, et al. DrugRep: an automatic virtual screening server for drug repurposing. Acta Pharmacol Sin. 2023;44(4):888-896.
CrossRef - Hubálek F, Binda C, Khalil A, et al. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. Journal of Biological Chemistry. 2005;280(16):15761-15766.
CrossRef - Odhar HA, Kareem SM, Alhaideri MRA, Hasan MA, Geldenhuys WJ. The design and evaluation of a novel monoamine oxidase b inhibitor through in silico approach. International Journal of Pharmaceutical Quality Assurance. 2019;10(2):349-355.
CrossRef - Odhar HA, Hashim AF, Humad SS. Molecular docking analysis and dynamics simulation of salbutamol with the monoamine oxidase B (MAO-B) enzyme. Bioinformation. 2022;18(3):304.
CrossRef - Morris GM, Ruth H, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30(16):2785.
CrossRef - Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J Chem Inf Model. 2021;61(8):3891-3898.
CrossRef - De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A. Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci U S A. 2005;102(36):12684-12689.
CrossRef - Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem. 2015;58(9):4066.
CrossRef - Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7.
CrossRef - Banerjee P, Kemmler E, Dunkel M, Preissner R. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024;52(W1):W513-W520.
CrossRef - Salo-Ahen OMH, Alanko I, Bhadane R, et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2021, Vol 9,. 2020;9(1):1-63.
CrossRef - Krieger E, Vriend G. YASARA View – molecular graphics for all devices – from smartphones to workstations. Bioinformatics. 2014;30(20):2981-2982.
CrossRef - Odhar HA, Kareem SM, Alhaideri MRA, Hasan MA, Geldenhuys WJ. The Design and Evaluation of a Novel Monoamine Oxidase B Inhibitor Through in Silico Approach. International Journal of Pharmaceutical Quality Assurance. 2019;10(2):349-354.
CrossRef - Zhang J, Xu X, Li N, et al. Licoflavone B, an isoprene flavonoid derived from licorice residue, relieves dextran sodium sulfate-induced ulcerative colitis by rebuilding the gut barrier and regulating intestinal microflora. Eur J Pharmacol. 2022;916.
CrossRef - Liu L, Geng X, Zhang J, Li S, Gao J. Structure-based discovery of Licoflavone B and Ginkgetin targeting c-Myc G-quadruplex to suppress c-Myc transcription and myeloma growth. Chem Biol Drug Des. 2022;100(4):525-533.
CrossRef - Aleixo de Carvalho LS, Geraldo RB, de Moraes J, et al. Schistosomicidal activity and docking of Schistosoma mansoniATPDase 1 with licoflavone B isolated from Glycyrrhiza inflata (Fabaceae). Exp Parasitol. 2015;159:207-214.
CrossRef - Lee S, Yun B, Kim M, et al. Phenolic compounds isolated from Psoralea corylifolia inhibit IL-6-induced STAT3 activation. Planta Med. 2012;78(9):903-906.
CrossRef - Chen CY, Chen CC, Shieh TM, et al. Corylin Suppresses Hepatocellular Carcinoma Progression via the Inhibition of Epithelial-Mesenchymal Transition, Mediated by Long Noncoding RNA GAS5. Int J Mol Sci. 2018;19(2).
CrossRef - Chen CC, Kuo CH, Leu YL, Wang SH. Corylin reduces obesity and insulin resistance and promotes adipose tissue browning through SIRT-1 and β3-AR activation. Pharmacol Res. 2021;164.
CrossRef - Gu J, Zhao L, Chen YZ, et al. Preventive effect of sanguinarine on intestinal injury in mice exposed to whole abdominal irradiation. Biomedicine and Pharmacotherapy. 2022;146.
CrossRef - Achkar IW, Mraiche F, Mohammad RM, Uddin S. Anticancer potential of sanguinarine for various human malignancies. Future Med Chem. 2017;9(9):933-950.
CrossRef - Ke Q, Duan K, Cheng Y, Xu S, Xiao S, Fang L. Sanguinarine Exhibits Antiviral Activity against Porcine Reproductive and Respiratory Syndrome Virus via Multisite Inhibition Mechanisms. Viruses. 2023;15(3).
CrossRef - Wangchuk P, Keller PA, Pyne SG, et al. A new protoberberine alkaloid from Meconopsissimplicifolia (D. Don) Walpers with potent antimalarial activity against a multidrug resistant Plasmodium falciparum strain. J Ethnopharmacol. 2013;150(3):953-959.
CrossRef - Park ES, Hwang YS, Ryu HW, et al. Paulownin elicits anti-tumor effects by enhancing NK cell cytotoxicity through JNK pathway activation. Front Pharmacol. 2024;15:1439079.
CrossRef - Škovranová G, Molčanová L, Jug B, et al. Perspectives on antimicrobial properties of Paulownia tomentosa Steud. fruit products in the control of Staphylococcus aureus infections. J Ethnopharmacol. 2024;321.
CrossRef - Kobayashi Y, Hoshikuma K, Nakano Y, Yokoo Y, Kamiya T. The positive inotropic and chronotropic effects of evodiamine and rutaecarpine, indoloquinazoline alkaloids isolated from the fruits of Evodia rutaecarpa, on the guinea-pig isolated right atria: possible involvement of vanilloid receptors. Planta Med. 2001;67(3):244-248.
CrossRef - Moon TC, Murakami M, Kudo I, et al. A new class of COX-2 inhibitor, rutaecarpine from Evodia rutaecarpa. Inflamm Res. 1999;48(12):621-625.
CrossRef - Kim CY, Lee GH, Lee SY, Bui ATN, Jeong HG. Rutaecarpine Protects Human Endothelial Cells from Oxidative-Stress-Induced Apoptosis via TRPV1- and AhR-Mediated Nrf2 Activation. Antioxidants (Basel). 2025;14(5).
CrossRef - Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3-26.
CrossRef - Oertel W, Schulz JB. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J Neurochem. 2016;139 Suppl 1:325-337.
CrossRef - Gaweska H, Fitzpatrick PF. Structures and Mechanism of the Monoamine Oxidase Family. Biomol Concepts. 2011;2(5):365.
CrossRef - Chinta SJ, Andersen JK. Redox imbalance in Parkinson’s disease. BiochimBiophys Acta Gen Subj. 2008;1780(11):1362-1367.
CrossRef - Hevener KE, Zhao W, Ball DM, et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model. 2009;49(2):444-460.
CrossRef - Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615-2623.
CrossRef - Di L, Kerns EH. Chapter 19 – Pharmacokinetics. In: Di L, Kerns EHBTDLP (Second E, eds. Second Edi. Academic Press; 2016:267-281.
CrossRef - Wang W, Kim MT, Sedykh A, Zhu H. Developing Enhanced Blood–Brain Barrier Permeability Models: Integrating External Bio-Assay Data in QSAR Modeling. Pharm Res. 2015;32(9):3055.
CrossRef - Gadaleta D, Vuković K, Toma C, et al. SAR and QSAR modeling of a large collection of LD50 rat acute oral toxicity data. J Cheminform. 2019;11(1):58.
CrossRef - Saikia S, Bordoloi M. Molecular Docking: Challenges, Advances and its Use in Drug Discovery Perspective. Curr Drug Targets. 2019;20(5):501-521.
CrossRef - Feig M, Nawrocki G, Yu I, Wang PH, Sugita Y. Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J Phys Conf Ser. 2018;1036(1):012010.
CrossRef - Milczek EM, Binda C, Rovida S, Mattevi A, Edmondson DE. The “gating” residues Ile199 and Tyr326 in human monoamine oxidase B function in substrate and inhibitor recognition. FEBS J. 2011;278(24):4860.
CrossRef - Li M, Binda C, Mattevi A, Edmondson DE. Functional role of the “aromatic cage” in human monoamine oxidase B: structures and catalytic properties of Tyr435 mutant proteins. Biochemistry. 2006;45(15):4775-4784.
CrossRef
List of Abbreviations
Å: Angstrom.
GI: Gastrointestinal.
IC50: Median inhibitory concentration.
LD50: Median lethal dose.
Log P: Logarithm of partition coefficient.
MAO: Monoamine oxidase.
MD: Molecular dynamics.
MM-PBSA: Molecular mechanic Poisson Boltzmann surface area.
PD: Parkinson’s disease.
PSA: Polar surface area.
RMSD: Root mean square deviation.
RO5: Rule of five.
TCM: Traditional Chinese Medicine.
VDss: Steady state volume of distribution.
Accepted on: 09-06-2026
Second Review by: Dr. Venkataramana Singamaneni and Dr. Amit Panaskar
Final Approval by: Dr. Jihan Seid Hussein




