
Manuscript accepted on :14-09-2024
Published online on: 02-10-2024
Plagiarism Check: Yes
Reviewed by: Dr. Ameer Ali Shakr Hadi and Dr. Prakash Balu
Second Review by: Dr. Althaf Hussain Basha SK
Final Approval by: Dr. Prabhishek Singh
Sarder Arifuzzaman1* , Zubair Khalid Labu1, Md. Harun -Or- Rashid1, Farhina Rahman Laboni1, Mst. Reshma Khatun2, Md Sajib Ali2, Shadek Hossain2 and Nargis Sultana Chowdhury2
, Zubair Khalid Labu1, Md. Harun -Or- Rashid1, Farhina Rahman Laboni1, Mst. Reshma Khatun2, Md Sajib Ali2, Shadek Hossain2 and Nargis Sultana Chowdhury2
1Department of Pharmacy, World University of Bangladesh, Uttara, Dhaka-1230, Bangladesh
2Department of Pharmacy, Manarat International University, Ashulia, Dhaka, Bangladesh
Corresponding Author E-mail:sarder.arifuzzaman@pharmacy.wub.edu.bd
DOI : https://dx.doi.org/10.13005/bpj/2960
Abstract
Studies have demonstrated the association between LXR activity dysregulation with many diseases, including atherosclerosis, diabetes and cancer. In recent years, several LXR agonists have surfaced, but none have been approved for human use due to adverse effects or unforeseen reasons. In this study, we first analysed the mRNA and protein expression of LXRs across tissues, network and pathway analysis, and reinterpreted their physiological function and disease association by utilizing multiple biological data repositories, including RNA-seq human protein atlas, DisGeNET, etc. Then, we performed ligand-based virtual screening, chemico-pharmacokinetic analysis, docking and simulation to identify potential new compounds. Our findings of mRNA, protein expression, network and disease enrichment analysis reveal diverse physiological functions of LXRs addressing the possibility of pharmacological manipulation with small molecules would provide therapeutic strategies for disease management. Evaluation of the docking and chemico-pharmacokinetic properties directed to the selection of LXR-623 and AZ876 as promising candidates for LXR-α and LXR-β for further in-silico investigation. Comprehensive screening for new ligands targeting LXRs based on the chemical structures of LXR-623 and AZ876, identified ZINC000005399501 and ZINC000021912941 with the highest binding affinity (−9.8 and −10.7 kcal/mol) for LXRα and LXRβ, respectively. Our results also supported in simulation study, along with favorable chemico-pharmacokinetic features.
Keywords
Agonists; Atherosclerosis; Chemico-Pharmacokinetic; Docking; LXRs; Metabolic Disease; Virtual Screening
Download this article as:| Copy the following to cite this article: Arifuzzaman S, Labu Z. K, Rashid M. H. O, Laboni F. R, Khatun M. R, Ali M. S, Hossain S, Chowdhury N. S. Identification of Novel Compounds Targeting the Liver X Receptor (LXR): In-silico Studies, Screening, Molecular Docking, and Chemico-pharmacokinetic Analysis. Biomed Pharmacol J 2024;17(3). |
| Copy the following to cite this URL: Arifuzzaman S, Labu Z. K, Rashid M. H. O, Laboni F. R, Khatun M. R, Ali M. S, Hossain S, Chowdhury N. S. Identification of Novel Compounds Targeting the Liver X Receptor (LXR): In-silico Studies, Screening, Molecular Docking, and Chemico-pharmacokinetic Analysis. Biomed Pharmacol J 2024;17(3). Available from: https://bit.ly/3Y99mxS |
Introduction
Liver X receptors (LXR), a member of the nuclear receptor family are important regulators of cholesterol, fatty acid, and glucose homeostasis.1 To date, two isoforms of LXRs have been discovered and are given the nuclear receptor nomenclature symbols NR1H3 (LXR-α) and NR1H2 (LXR-β).1 The well-established role of LXRs was confirmed and reaffirmed by the published data on LXRs in gene-disease association, network, mRNA and protein expression in tissues.2-4 LXRs have been shown to function as direct transcriptional regulators for genes involved in cholesterol and lipid metabolism regulation, including ATP Binding Cassette transporter (ABC), Apolipoprotein E (ApoE), CholEsterylester Transfer Protein (CETP), Fatty Acid Synthase (FAS), cholesterol 7α-hydroxylase (CYP7A1), sterol regulatory element binding protein 1c (SREBP-1c), and stearoyl-CoA desaturase-1 (SCD-1). There are reports LXRs activation increases hepatic VLDL production by 2.5-fold, and produces large TG-rich VLDL particles in the liver.5-8 LXR accelerates the conversion of cholesterol to bile acids, lowers the amount of cholesterol at the cellular level, and enhances reverse cholesterol transport to the liver.9 Thus, evidences of links between LXR dysregulations and the onset of metabolic diseases (e.g., hyperlipidemia,atherosclerosis)9,10 attracted researchers to the innovation of ligands targeting LXRs.
In recent years, several LXR agonists have been studied in preclinical trials with the intent to develop new drugs for atherosclerosis, diabetes, anti-inflammation, Alzheimer’s disease, and cancer.11-15 Among these reported LXR agonists, T0901317, LXR-623 and GW3965 showed efficacy to reduce cholesterol levels not only in the serum but also in the liver of mice with various diseases.15-18 A recent study demonstrated that SR9243 reduces intrahepatic inflammation caused by nonalcoholic steatohepatitis and regulates lipid metabolism in cancer cells.19,20 In animal models, GSK3987, an agonist of pan LXR-α/β with EC50s of 40–50 nM, decreases triglyceride buildup and increases cellular cholesterol efflux.21 There are also LXR agonists that have been tested in clinical trials, including LXR-623, BMS-779788, BMS-852927, and AZ876.22-25 Almost all the agonists have been abandoned owing to ineffectiveness or serious side effects in clinical trial.22-25 There is an unmet need to develop the most suitable LXR-α/β selective agonists with fewer side effects, as none of these compounds pass the clinical trial due to ineffectiveness or substantial side effects.
Molecular docking and molecular dynamic (MD) simulation are two of the most popular, quick, affordable, and straightforward computational computer-assisted methods for designing or finding small molecules for drug development.26-31 Docking study is often employed in drug discovery to predict the binding affinity and binding mode of a small molecule (ligand) with a target protein or nucleic acid.26-28,32,33 While MD simulation provides insights into the dynamical behavior of biomolecules including the motion of atoms and molecules over time.31,34 Combining these techniques provides a more comprehensive understanding of the ligand-receptor interaction, accounting for flexibility and dynamic changes. To confirm the understandings and determine the true therapeutic potential of the research, a preclinical or clinical experimental endorsement is required.
A web-based virtual screening tool called SwissSimilarity (http://www.swisssimilarity.ch/) allows quick screening of pharmaceuticals, bioactive small compounds, and commercially available ligands from PubChem, the ZINC database, Drug Bank, and other sources.35 SwissSimilarity employs molecular fingerprints and fast nonsuperpositional or superpositional 3D shape similarity techniques.35 In addition to SwissSimilarity, another popular web tool for calculating physicochemical attributes, pharmacokinetics assessment, drug-likeness, and medicinal chemistry friendliness of small compounds is SwissADME (http://www.swissadme.ch/).36 Important characteristics of SwissADME are Lipinski’s rule of five, certain pharmacokinetic factors to help with the early phases of the drug discovery process, and forecasts of properties of drug absorption, distribution, metabolism, and excretion (ADME).37,38
Therefore, the goal of this work was to find ligands that have the maximum affinity to the target LXRs by using chemico-pharmacokinetic methods, molecular docking, virtual screening, in-silico research, and MD modelling. In addition to being underutilized in clinical practice at the moment, these ligands have less chance of side effects and may be used to treat cancer, atherosclerosis, and metabolic disorders. Furthermore, a thorough investigation was carried out using publically accessible databases to reevaluate the levels of mRNA and protein expression of LXR-α and LXR-β in both healthy and diseased tissues. This allowed for a revision of the associations between these proteins and disorders.2-4,39,40 Ligands with substantial binding affinities and specificities for LXR can be found and considered as viable candidates for additional research by adjusting the interaction between the drug candidate and the LXR target protein.
Materials and Methods
In-silico study of mRNA and protein expression of LXRs
We performed an in-silico LXRs’ mRNA and protein expression study to learn more about their physiological function with gene IDs 10062 and 736 for LXR-α and LXR-β, respectively. We curated the mRNA expression data for LXR-α and LXR-β from the HPA RNA-seq database (https://www.ncbi.nlm.nih.gov/gene/) of normal tissues.40 The data are shown in Figure 1a. Additionally, we observed the protein level expression using data from the Human Protein Atlas (https://www.proteinatlas.org/). 4,39 Figure 1b displays the curated protein expression data for LXR-α and LXR-β.
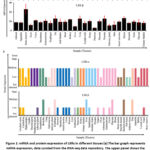 |
Figure 1: mRNA and protein expression of LXRs in different tissues |
Gene-disease Association Analysis
To observe major diseases involving these genes, we performed gene-disease association (GDA) analysis for LXRs using the web-based platform DisGeNET v7.0 (https://www.disgenet.org/), one of the largest publicly accessible collections of genes and variants linked to human diseases.3 DisGeNET combines information from expert-curated repositories, GWAS catalogues, animal models, and scientific literature. DisGeNET employs a text-mining technique to prioritise the genotype-phenotype associations.3 The GDA analysis data are shown in Figure 2a.
Analysis of gene networks and pathways
To obtain further insight into the physiological and pathological significance of LXR expression, we conducted a protein-protein interaction analysis, specifically, we examined the interaction using IntAct (https://www.ebi.ac.uk/intact/). To address LXR involvement in biological/physiological pathways, we performed pathway enrichment analysis using WebGestalt (https://www.webgestalt.org/), a functional enrichment analysis web tool.2 WebGestalt follows well-established and complementary methods for enrichment analysis, including overrepresentation analysis.2
Target protein identification and selection
Clinical data has shown that LXRs play a significant role in controlling glucose, fatty acid, and cholesterol homeostasis. Additionally, LXR agonists have been shown to be successful in treating mouse models of cancer, anti-inflammation, atherosclerosis, and diabetes. The aforementioned proteins are highly essential in the overall management of atherosclerosis, diabetes, anti-inflammation, Alzheimer’s disease, and cancer due to the complexity of LXRs’ role in the pathophysiology and the promising outcomes of using LXR agonists. They are also suitable for in silico studies. Therefore, the target proteins for the current molecular docking investigation are LXR-α and LXR-β, whose PDB IDs are 1uhl and 5ajy, respectively. Ligands chosen according to their affinity for these structures have been redocked for both target LXRs.
Ligand preparation for docking
Chemical ligands were identified through a literature search: T0901317, AZ876, BMS-779788, BMS-852927, GSK3987, LXR-623, SR9243, GW3965, 24S-hydroxycholesterol, 24R-hydroxycholesterol, and 5,6-epoxycholesterol.18-20,22,25,41,42 Using MarvinSketch, a ChemAxon’s desktop programmes for chemistry programme for sketching and visualising chemical structures, the structures were reprocessed in PDB or Mol2 format. We added hydrogen atoms for the compounds lacking hydrogen atoms and defined rotatable bonds that will be used for flexible docking. The ligands obtained from a database screening were selected for creating a new structure using ChemAxon software. We cleaned the ligand structure by removing any unwanted atoms or molecules, such as solvent molecules or counter ions. Generated the 3D coordinates for the ligands using the same software or web tool. The ligand structures are optimized by minimizing the energy using a molecular mechanics force field. We saved the ligand structure in a format that is compatible with the docking software, such as PDB or Mol2 format.
Target protein preparation for Docking
The target protein sequencefor LXR-α (1uhl) and LXR-β (5jy3) was obtained from the PDB online platform (https://www.rcsb.org/). Then we selected and deleted the ligands and water (LXR-α/β-drug) from the complex using PyMOL2.5 software,43,44 because ligand-free and water-free cocrystallized 3D protein structure is needed for molecular docking assay.45 A molecular insertion study was conducted to identify the binding sites between the target protein and different chelating agents. We saved the target protein structure in a format that is compatible with the docking software, such as PDB format. In order to identify the binding pockets in the chosen target proteins where the ligands are most likely to bind with stable free energy, we used the web applications 3DligandSite and COACH-D.46,47
Screening of compound libraries
For LXR-α and LXR-β, ligands with comparable chemical structures and therapeutic potential were screened using the online tool SwissSimilarity (http://www.swisssimilarity.ch/). SwissSimilarity allows screening a wide range of compound libraries such as DrugBank, ChEMBL, LigandExpo, ZINC, and many more.35 The SMILES format was used to submit the query molecule. These ligands were identified using the chemical structures of the compounds that showed the highest affinity for LXR-α and LXR-β during docking tests. The ZINC provided the ligands in SDF format for download.48 The format was changed using OpenBabelGUI 3.0.1 to be compatible with AutoDock 4 and AutoDock Vina. The rotatable bonds were then identified and the required charges were added.45
Molecular Docking Analysis
We conducted molecular docking analysis using PyRx (https://pyrx.sourceforge.io/), a virtual suite programme for Computational Drug Discovery.49 PyRx is built with many established open source software including AutoDockTools to generate input files, python as a programming/scripting language, wxPython for cross-platform GUI, the Visualization ToolKit, Enthought Tool Suite including Traits for application building blocks, Open Babel for importing SDF files and removing salts and energy minimization, matplotlib for 2D plotting, AutoDock 4 and AutoDock Vina for docking.49 We uploaded the ligand and protein files to the PyRx interface as mol2 and pdb files, respectively. Protein and ligand files were again cleaned and the structures processed to be devoid of unlikeliness such as missing atoms, protonation states, and water molecule removal before running the docking analysis. Here, we used Open Babel to create a pdbqt file (protein.pdbqt, ligand.pdbqt) for the ligand and protein files. To facilitate the docking calculations a 3D grid around the target binding site in the receptor is created using AutoGrid. The grid box dimensions were set x,y,z dimension, and the center grid box coordinates x, y, z center, respectively. In this study, the grid box dimensions were set x,y,z as 55, 55, 55 Å, and the coordinates of center x, y, and z were 44.53, -2.4883, and 21.7037 Å, respectively.
The binding affinity between the ligand and receptor is estimated by the Autodock Vina docking algorithm. The Autodock Vina uses scores each ligand poses and ranks the poses to identify potential binding candidates. The scoring function in AutoDock Vina calculates intermolecular interactions such as van der Waals forces, hydrogen bonding, steric and electrostatic interactions between the ligand and the protein, and the desolvation energy. Once the calculations are done, results will be populated as seen in the below table with the Binding Affinity (kcal/mol) values. The more negative the numerical values for the binding affinity, the better the predicted binding between a ligand and a protein. In the context of docking evaluation, the RMSD is computed relative to the native ligand to ascertain how well the anticipated posture approaches the crystallographic pose. When it comes to computational molecular docking, algorithms are considered legitimate and dependable if they generate poses with RMSD values less than 2 Å, where RMSD is determined to the native ligand. A lower RMSD value indicates better accuracy of the docking technique and a better fit between the anticipated and experimental positions49. A validation of the results is performed using experimental data or by comparing with known binding modes. Docking results are visualized into 2D and 3D images by Pymol and Biovia Discovery Studio, both are powerful molecular visualization tools and are used for docking analysis to visualize protein-ligand interactions.
Validation of docking method
A commonly used strategy is called “pose selection,” which entails re-docking a chemical with a known conformation and orientation—usually from a co-crystal structure—into the target’s active site using docking algorithms. When programmes can return poses with a RMSD value from the known conformation—which, depending on ligand size, is frequently 1.5 or less than 2 Å—they are considered effective.
Chemico-Pharmacokinetic Profile Predictions
Using the online technology provided by SWISSADME (http://www.swissadme.ch/), we evaluated the pharmacokinetics, drug-likeness, and medicinal chemistry compatibility of small molecules that were identified as the most promising candidates for LXR-α and LXR-β through molecular docking and virtual screening. The ligands were filtered based on Lipinski’s “Rule of Five,” which encompasses criteria such as molecular weight (MW) being less than 500, log P being less than 5, the number of hydrogen bond donors being less than 5, and the number of hydrogen bond acceptors being less than 10. Additionally, we employed established ADME pharmacokinetics prediction methods, including assessment of aqueous solubility (PlogS), blood/brain permeability (PlogBB), intestinal barrier permeability (logHIA), cell permeability (PCaco-2), substrate/non-inhibitor status (logPgp), as well as cell permeability (LogPapp) and CYP inhibition, to identify the most suitable ligands for LXR-α and LXR-β targeting. The query molecules were submitted in SMILES format.
Molecular dynamics simulation
Further assessment of the binding stabilities of these putative LXR-α/β-agonists is done using molecular dynamics simulation with QwikMD and NAMD (https://www.ks.uiuc.edu/Research/qwikmd/). Using the SwissParam-generated ligand force field and the CHARMM general force field for proteins, the structure of the ligand–receptor complex in the optimal docking position was simulated. The simulation was run with an implicit solvent model and a salt concentration of 0.15 M. We present the results of a 50 ns production simulation of the ligand–LXR-α/β complexes using the VMD1.9.4a53 toolset. To analyze the results of the simulation in QwikMD, in the simulation setup, we load the corresponding .qwikmd file. The pop-up menu provides the option to load any generated trajectory (e.g., equilibration, production, etc.), from which we select the “production” option and specify a trajectory frame step. For equilibration simulation we set a temperature of 60 K to 300 K at 1.242-second frequency and production simulation we set a constant temperature of 300 K. The 2.5 ps frequency at which the simulation results were saved was used. During the examination of the simulations, RMSF and RMSD values for the ligand–LXR-α/β complexes were documented. To ensure greater precision MD simulation was repeated twice for each complex, and the average outcome was employed for analysis. We also determine the nonbond interaction energy (kcal/mol) between the ligand and protein. Frame 0 serves as the reference frame in RMSD analysis, and each frame’s structure is aligned with frame 0’s to measure real structural fluctuations that occur throughout the simulation.
Results
Many physiological processes and disorders are regulated by LXRs, as evidenced by mRNA and protein expression, gene-disease association, network, and pathway enrichment analysis.
Using the publicly accessible RNA-seq databases (https://www.ncbi.nlm.nih.gov/gene/) and the human protein atlas (https://www.proteinatlas.org/), we initially looked at both mRNA and protein expression across organs.4,39,40 LXR-α expression is restricted to the liver, kidney, gut, fat tissue, lung, and spleen and is predominantly detected in fat. LXR-β is expressed in almost all tissues and organs (Figure 1a, b). While LXR-α/β show overlap in a number of tissues, their tissue distribution patterns diverge greatly. The divergent expression patterns suggest that LXR-α/β has different functions in regulating physiological processes. Based on DISGENET’s gene-disease association analysis, LXR-α has been associated with several metabolic disorders, including atherosclerosis, coronary heart disease, metabolic syndrome, type 2 diabetes, and coronary hyperlipidemia.3 LXR-β has also been connected to a number of metabolic diseases as well as cancer (Figure 2a).
Based on network analysis, it was projected that LXRs physically associate or directly interact with many proteins such as transporters (PPARA), transcription factors (RXRG, RXRA, RXRB, NCOR1), enzymes (KDM1A, SUV39H1), and other essential proteins (EDF1, MDFI, CORO2A) (Figure 2b). It is commonly recognised that these transporters, transcription factors, and enzymes regulate lipids and cholesterol. WebGestalt’s (https://www.webgestalt.org/) pathway enrichment study also identified the main pathways implicated in LXRs. Nuclear receptors in lipid metabolism and toxicity, cholesterol and lipid homeostasis, cholesterol-derived oxysterols, the PPAR alpha route, the development of white fat cells, and the PPAR signalling pathway are among the pathways (Figure 2c). All of these findings point to the possibility that small molecules can interfere in atherosclerosis through pharmacological LXR activation.
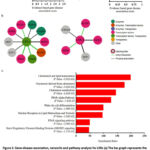 |
Figure 2: Gene-disease association, networks and pathway analysis for LXRs |
Molecular docking analysis targeting LXR-α for the selected ligands shows distinct binding affinities and patterns of interaction with amino acids
The 3D structure of the ligands was obtained from the literature search. Both the ligands and active sites of the protein were prepared carefully to meet the requirements of the docking software. Our docking analysis results demonstrated the binding affinity of ligands within the binding pocket and the interacting amino acid for AZ876, BMS-779788, BMS-852927, GSK3987, GW3965, LXR-623, SR9243, T0901317, 24(S)-hydroxycholesterol and 22(R)-hydroxycholesterol (Figure 3 and Table 1). Out of all the ligands investigated, LXR-623 had the highest binding affinity for LXR-α, indicating the most potential for a robust interaction with LXR-α.
Our results showed the interacting amino acids for T0901317 were GLU308, VAL311, GLN313, SER383, ARG387, ILE389, and LYS435 while 24R-hydroxycholesterol, having the lowest docking score (-6.5 kcal/mol), showed interaction with ARG387, ILE389, ARG443. The ARG387 and GLN313 illustrated traditional hydrogen bonds, halogen (fluorine) bonds were represented by GLU308, SER383, and VAL311, and Pi-cation and Pi-anion interactions were shown by LYS435 in the LXR-α-T0901317 complex within 4 Å. The ILE389 demonstrated Pi-alkyl interaction, while the GLU312, HIS386, LEU383, and ASP390 displayed vander waals interaction in the LXR-α-T0901317 complex.
AZ876 interacts with THR292, ARG369, PRO370, ASN371, ARG415, TRP443, and ASP444 and has an affinity of −8.5 kcal/mol for LXR-α. BMS-779788 and BMS-852927 interact with PHE229, LEU260, VAL263, SER264, ASP318 ILE370, and ARG373, ALA374, LYS452, ARG497, LEU501, LEU504, PHE503, ALA528, with binding affinities of − 8.3 and 8.2 kcal/mol, respectively. GSK3987 binds to SER366, VAL386, GLU387, HIS390, PHE404, LEU408, LEU411, and ARG415. Its binding affinity is −7.4 kcal/mol. GW3965 interacted with amino acids ARG226, PHE229, VAL263, ARG305, and TYR306, with an affinity of -7.0 kcal/mol. Although LXR-623 had a −10.6 kcal/mol affinity for LXR-α, it also binds to LYS452, ARG497, GLU378, ALA374, ASN377, MET525 and ARG373.
 |
Figure 3. Binding models of LXR–α agonist complexes. |
Table 1: The binding affinity of ligands within the binding pocket and the amino acids that interact with LXR-α.
|
Ligand |
Binding Affinity (Kcal/mol) |
rmsd/ub |
rmsd/lb |
Interacting amino acid |
|
LXR-623 |
-10.2 |
2.153 |
1.402 |
GLU308, VAL311, GLN313, SER383, ARG387, ILE389, LYS435 |
|
AZ876 |
-8.5 |
5.52 |
2.891 |
ILE370, ARG373, ALA374, TRP376, LYS452, LEU504, PHE508 |
|
BMS-779788 |
-8.3 |
1.746 |
1.312 |
GLU308, GLU312, HIS386, ARG387, ILE389, LEU396, LYS435 |
|
BMS-852927 |
-8.2 |
30.743 |
26.208 |
ILE370, ARG373, ALA374, LYS452, ARG497, LEU501, LEU504, PHE503, ALA528 |
|
GSK3987 |
-7.4 |
8.99 |
3.51 |
ILE370, ARG373, ALA374, GLU378, LEU501, LEU504, ALA528 |
|
T0901317 |
-7.3 |
4.489 |
2.429 |
ARG373, ALA374, ASN377, GLU378, LYS452, ARG497, LEU501, LEU504, MET525 |
|
SR9243 |
-7.2 |
10.006 |
3.63 |
GLU312, HIS386, ARG387, ILE389, LEU396, LYS435, LEU438 |
|
GW3965 |
-7.0 |
4.685 |
2.761 |
ARG373, ALA374, ASP450, ARG497, LEU504 |
|
24S-hydroxycholesterol |
-6.9 |
5.959 |
4.397 |
ILE445, TYR468, LEU490, LEU493, PRO494 |
|
24R-hydroxycholesterol |
-6.5 |
2.06 |
1.466 |
ARG387, ILE389, ARG443 |
RMSD (root mean square deviation), rmsd/lb (root mean square deviation/upper bound), rmsd/lb(root mean square deviation/lower bound) are calculated by the AutoDock Vina of PyRx tool (total runs=9).
Molecular docking analysis targeting LXR-β for the selected compounds shows distinct binding affinities and patterns of interaction with amino acids
LXR-β complexed with BMS-852927 (PDB ID 5jy3) was redocked, binding affinities and the RMSD were computed for AZ876, BMS-779788, GSK3987, GW3965, LXR-623, SR9243, T0901317, 24(S)-hydroxycholesterol and 22(R)-hydroxycholesterol, following a similar protocol to that for LXR-α. Table 2 displays the affinities for LXR-β that all of the chosen compounds have shown. Of the ligands that were studied, AZ876 interacts with the amino acids PHE268, PHE271, THR271, LEU274, ALA275, SER278 MET312, LEU313, GLU315, THR316, ARG319, PHE329, PHE340, LEU345, PHE349, ILE 350, ILE353, HIS435 and had the highest affinity of −10.8 kcal/mol for LXR-β (Figure 3, Table 2). The interacting residues involved in the formation of vander waals forces, hydrogen bond, pi-sigma, pi-sulfur, amide-pi-stacked, alkyl, and pi-alkyl interactions in the LXR-β-AZ876 complex within 2.4 Å. Although van der Waals force was demonstrated by SER278 LEU313, GLU315, THR316, PHE340, PHE349, and ILE350, PHE271 showed traditional hydrogen bonds; LEU274 represented the carbon-hydrogen bond; alkyl and pi-alkyl were demonstrated by PHE268, ALA275, PHE329, LEU345, ILE353, HIS435, pi-sigma by ILE353, and pi-sulfur by ARG313, MET312.
The compound BMS-779788, which interacts with PHE271, ALA275, ILE309, MET312, PHE329, PHE340, LEU345, and HIS435; and BMS-852927, which interacts with PHE271, ALA275, LYS305, ILE309, MET312, PHE329, PHE340, LEU345, HIS435, and SER436; both have lower affinity than SR9243, 22R-hydroxycholesterol, GW3965, LXR-623, T0901317 and GSK3987 (Table 2). GSK3987 has a binding affinity of -9.8 kcal/mol and binds to the ALA275, LEU345, and HIS435. GW3965 and LXR-623 had a similar affinity of -9.8 kcal/mol, despite the differences in the amino acid interactions. In LXR-623, the amino acids that interacted were PHE268, LEU274, ALA275, LEU345, ILE353, and HIS435, whereas in GW3965, they were PHE271, LEU274, ALA275, MET312, PHE329, PHE340, LEU345, and HIS435. GSK3987 has a binding affinity of -9.8 kcal/mol and binds to the ALA275, LEU345, and HIS435.
SR9243 and T0901317 bind to the amino acid PHE268, SER274, ALA275, ILE309, MET312, LEU313, PHE329, LEU330, LEU345 and ARG319, THR316, GLU281, PHE329, LEU330, LEU345, ILE353, with affinities of -10.3 and -9.7 kcal/mol, respectively. Despite being isomers, 24R- and 24S-hydroxycholesterol displayed varying levels of binding affinity. The binding affinity of 24R- and 24S-hydroxycholesterol were -9.8 and 8.1 kcal/mol, respectively. In relation to 24R-hydroxycholesterol, the interacting amino acids were LEU274, ALA275, SER278, PHE 329, LEU345, and HIS435; in contrast, PHE268, ALA275, MET312, PHE329, LEU274, LEU345, ILE353 and HIS435 were associated with 24S-hydroxycholesterol.
 |
Figure 4: Binding models of LXR–β agonist complexes. |
Table 2: The binding affinity of ligands within the binding pocket and the amino acids that interact with LXR-β.
|
Ligand |
Binding Affinity (Kcal/mol) |
rmsd/ub |
rmsd/lb |
Interacting amino acid |
|
AZ876 |
-10.8 |
1.625 |
1.734 |
PHE268, PHE271, LEU274, ALA275, MET312, PHE 329, LEU345, ILE353, HIS435 |
|
SR9243 |
-10.3 |
2.122 |
1.466 |
PHE268, SER274, ALA275, ILE309, MET312, LEU313, PHE329, LEU330, LEU345 |
|
22R-hydroxycholesterol |
-9.8 |
1.731 |
1.857 |
LEU274, ALA275, SER278, PHE 329, LEU345, HIS435 |
|
GW3965 |
-9.8 |
2.702 |
1.642 |
PHE271, LEU274, ALA275, MET312, PHE329, PHE340, LEU345, HIS435 |
|
LXR-623 |
-9.8 |
1.387 |
1.766 |
PHE268, LEU274, ALA275, LEU345, ILE353, HIS435 |
|
T0901317 |
-9.7 |
3.589 |
2.528 |
PHE272, SER274, ALA275, GLU281, MET312, THR316, ARG319, PHE329, LEU330, LEU345, ILE353 |
|
GSK3987 |
-9.2 |
3.57 |
2.39 |
ALA275, LEU345, HIS435 |
|
BMS-779788 |
-9.0 |
2.213 |
2.81 |
PHE271, ALA275, ILE309, MET312, PHE329, PHE340, LEU345, HIS435 |
|
BMS-852927 |
-8.9 |
3.09 |
2.356 |
PHE271, ALA275, LYS305, ILE309, MET312, PHE329, PHE340, LEU345, HIS435, SER436 |
|
24S-hydroxycholesterol |
-8.1 |
2.714 |
2.472 |
PHE268, LEU274, ALA275, MET312, PHE329, LEU345, ILE353, HIS435 |
RMSD (root mean square deviation), rmsd/lb (root mean square deviation/upper bound), rmsd/lb(root mean square deviation/lower bound) are calculated by the AutoDock Vina of PyRx tool (total runs=9).
Utilization of virtual screening to find new compounds that target LXR-α and LXR-β
Using the SwissSimilarity (http://www.swisssimilarity.ch/) online platform, a virtual search was done in the ZINC online database to find new compounds that target LXR-α and LXR-β. SwissSimilarity provides a diverse range of small molecule databases that can be used for screening purposes. These databases encompass drugs and clinical candidates, bioactive compounds, commercially available compounds, and synthesizable molecules. Various molecular fingerprints/vectors, which can either represent the 2D molecular structure or the 3D conformation of a compound, are employed by SwissSimilarity to identify structures that bear similarity to the query compound. The 2D methods involve the application of path-based FP2 fingerprint, extended-connectivity fingerprint with diameter 4, MinHash fingerprints, 2D pharmacophore fingerprints, and extended reduced graph fingerprints. Conversely, the 3D methods encompass Electroshape 5D vectors and extended 3D fingerprints.
SwissSimilarity analysis was employed to discover new compounds that target LXR-β. This search identified 400 ligands based on the configuration of AZ876, with similarity scores ranging from 0.94 to 0.354 (Supplementary file S1). Based on the structure of LXR-623, we identified 265 compounds in our inquiry with similarity scores ranging from 0.767 to 0.196 (Table 3 and Supplementary file S2). Figure 5 presents the 2D structure of the compounds identified for LXR-α in the ZINC database and figure 6 displays the 2D structure of the ten most significant molecules identified and screened for LXR-β in the ZINC database. A compound is completely distinct if its score is 0, and it is identical if it is 1 according to the SwissSimilarity study.
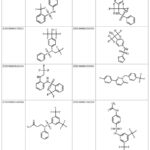 |
Figure 5: The most promising ligands identified for LXR-Α in the ZINC database |
Table 3: The similarity score obtained from swisssimilarity (https://www.swisssimilarity.ch/), binding affinity within the binding pocket and interacting amino acids of new ligands with LXR-α.
|
Ligand |
Similarity score |
Binding Affinity (Kcal/mol) |
rmsd/ub |
rmsd/lb |
Interacting amino acid |
|
ZINC000001550221 |
0.985 |
‒10.2 |
2.153 |
1.402 |
ARG373, ALA374, ASN377, GLU378, LYS452, ARG497, LEU501, LEU504, MET525 |
|
ZINC000000985503 |
0.926 |
‒9.8 |
5.52 |
2.891 |
GLU312, HIS386, ARG387, ILE389, LEU396, LYS435, LEU438 |
|
ZINC000058101934 |
0.837 |
‒10.13 |
1.746 |
1.312 |
ARG373, ALA374, ASP450, ARG497, LEU504 |
|
ZINC000095464663 |
0.817 |
‒12.3 |
3.743 |
2.208 |
ILE445, TYR468, LEU490, LEU493, PRO494 |
|
ZINC000003243391 |
0.709 |
‒9.6 |
8.99 |
3.51 |
GLU378, ARG464, TYR468, PHE486, LEU490, LEU491, PRO494 |
|
ZINC000016130131 |
0.637 |
‒8.2 |
4.489 |
2.429 |
ARG373, ALA374, ASN377, GLU378, LYS452, ARG497, LEU501, LEU504, MET525 |
|
ZINC000001042265 |
0.568 |
‒7.4 |
1.746 |
1.312 |
ARG373, ALA374, ASP450, ARG497, LEU504 |
|
ZINC000031669066 |
0.526 |
‒7.3 |
3.743 |
2.208 |
ILE445, TYR468, LEU490, LEU493, PRO494 |
Molecular docking analysis of the novel compounds targeting LXR-α and LXR-β show distinct binding affinities and patterns of interaction with amino acids
Among the identified novel compounds, ZINC000005399501 presented the highest binding affinity for LXR-α as determined by docking analysis.The binding affinity of the novel compounds identified targeting LXR-α ranges from -12.3 to -7.3 Kcal/mol as shown in Table 3. ZINC000005399501 interacts with ARG387, ILE389, ASP390, LYS435, LEU438, GLU308, and GLU312, according to the 2D study of the LXR-α-ZINC000005399501 complex. The interactions involved were van der Waals, alkyl, pi-alkyl, amide-pi stacked, conventional hydrogen bond, and carbon-hydrogen bond. The predominant interaction observed was the conventional hydrogen bond, followed by pi-sulfur, amide-pi stacked, alkyl, and pi-alkyl interactions involving GLU312, ILE389, LYS435, and LEU438. Fluorine and benzene are commonly used to enhance the properties of chemical compounds and displayed interaction with ARG387, ILE389, GLU308, SER383, HIS386, and GLU312. Specifically, the N‐(2‐((1R)‐2,2,2‐trifluoro‐1‐hydroxyethyl) phenyl group interacts with GLU308 and SER383, the benzene group interacts with GLU312, ARG387 and ILE389, and the sulfonamide group interacts with HIS386 and LYS435 (Figure 6).
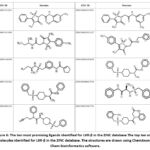 |
Figure 6: The ten most promising ligands identified for LXR-β in the ZINC database |
The 2D configuration, the similarity score, and the affinity towards LXR-β resulting from the process of docking for the ten most promising compounds that have not been utilized in pharmacotherapies are visually represented in Figure 7. The compound ZINC000021912941 demonstrated the uppermost binding affinity for LXR-β in the current virtual screening investigation, indicating its potential as a viable candidate for the development of a drug targeting LXR-β due to its potentially effective interaction with the target (as depicted in Figure 7 and Table 4). Previous reports in the scientific literature have documented that the binding affinities of compounds targeting LXR-β, as determined through in silico studies, fall within a range of -8.1 kcal/mol to -10.8 kcal/mol. In the current examination, it is noted that the binding affinity of all the top ten molecules falls within this typical range commonly observed in scientific publications (as illustrated in Figure 8 and Table 4). Notably, the compound ZINC000021912941 exhibits the highest affinity for LXR-β and forms interactions with the amino acids PHE271, MET312, PHE329, LEU345, and HIS435 (depicted in Figure 7). While the N-(2,3-dimethylphenyl) group interacts with the amino acids PHE271, LEU345, and HIS435, the 2,3-dihydro-1 lambda6,2-thiazol group in the compound contributes only one interaction with the protein (shown in Figure 8).
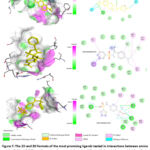 |
Figure 7: The 2D and 3D formats of the most promising ligands tested in interactions between amino acids of LXR-α. |
Table 4: The similarity score obtained from swisssimilarity (https://www.swisssimilarity.ch/), binding affinity within the binding pocket and interacting amino acids of new ligands with LXR-β.
|
Ligand |
Similarity score |
Binding Affinity (Kcal/mol) |
rmsd/ub |
rmsd/lb |
Interacting amino acid |
|
ZINC000021912941 |
0.96 |
‒10.7 |
2.122 |
1.466 |
PHE272, SER274, ALA275, GLU281, MET312, THR316, ARG319, PHE329, LEU330, LEU345, ILE353 |
|
ZINC000021912951 |
0.795 |
‒10.2 |
1.625 |
1.734 |
PHE268, LEU274, ALA275, LEU345, ILE353, HIS435 |
|
ZINC000021913098 |
0.787 |
‒10.03 |
1.325 |
1.724 |
LEU274, ALA275, LEU345, ILE353, |
|
ZINC000021913127 |
0.600 |
‒9.8 |
1.731 |
1.857 |
ALA275, LEU345, HIS435 |
|
ZINC000095446598 |
0.496 |
‒9.8 |
2.702 |
1.642 |
PHE271, ALA275, ILE309, MET312, PHE329, PHE340, LEU345, HIS435 |
|
ZINC000036398658 |
0.485 |
‒9.5 |
1.625 |
1.734 |
PHE268, LEU274, ALA275, LEU345, ILE353, |
|
ZINC000037207255 |
0.48 |
-9.2 |
2.531 |
1.257 |
PHE272, LEU274, ALA275, LEU345, ILE353, |
|
ZINC000040555976 |
0.47 |
-8.7 |
1.724 |
1.625 |
PHE268, LEU274, ALA275, LEU345, ILE353, |
|
ZINC000014043132 |
0.447 |
-9.1 |
1.857 |
1.325 |
GLU281, MET312, THR316, ARG319, PHE329, LEU330, LEU345, ILE353 |
|
ZINC000019867701 |
0.443 |
-8.9 |
1.642 |
1.731 |
SER274, ALA275, GLU281, MET312, THR316, ARG319, PHE329 |
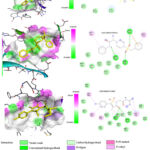 |
Figure 8: The 2D and 3D formats of the most promising ligands tested in interactions between amino acids of LXR-β |
In silico evaluation of the chemico-pharmacokinetic profile of the known and newly identified compounds
In the process of finding new drugs, it is vital to anticipate the pharmacokinetic characteristics, medicinal chemistry, druglike nature, and ADME parameters of one or more small molecules by computing physicochemical descriptors. We utilized SwissADME to evaluate the chemico-pharmacokinetic characteristics of newly discovered and existing candidates in silico. Table 5-9 and Supplementary file S3-S5 display the results of the chemico-pharmacokinetic parameters that were determined after the data was processed and entered into the SwissADME system. The molecular weight of the 8 molecules identified by Swissimilarity for LXR-α were less than 500 Da, there are less than five hydrogen-bond donors or less than ten hydrogen-bond acceptors (nitrogen and oxygen atoms), the computed logP (ClogP) is less than 5. They are either inhibitors or non-inhibitors of five major isoforms (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4) of the cytochrome P450 enzyme systems. We observed all 8 molecules are not only non-permeable to the blood-brain barrier but also low in GI absorption, and non-substrate of p-gp (Supplementary file S4). Similar to LXR-α, the 400 molecules found by Swissimilarity for LXR-β had chemicopharmacokinetics characteristics that were smaller than 500 Da. The H-bond acceptor and donor scores were 3, and 9, respectively. The consensus ClogP is less than 5, our findings revealed a variety of characteristics of these ligands in relation to the cytochrome P450 enzyme systems (Supplementary file S5).
The molecular weight of LXR-623, ZINC000005399501, AZ876 and ZINC000021912941 were 481.33 399.31, 439.57 and 414.43 Da, respectively, which reflects their suitability for oral drug development (Table 5). These compounds not only meet Lipinski’s criteria of molecular mass of less than 500 Da but also have no more than 5 hydrogen bond donors, no more than 10 hydrogen bond acceptors, and calculated implicit log P (iLOGP) that does not exceed 5 (or MlogP>4.15) (Table 5). In addition to Lipinski’s criteria, the MR (molecular refractivity) from 40 to 130 indicates additional features that increase druglikeness, and the TPSA (topological polar surface area) of a molecule greater than 1.40 nm2 tends to be poor at permeating cell membranes, while TPSA less than 0.90 nm2 is usually needed to penetrate the blood–brain barrier. Thus, except ZINC000021912941, all LXR-623, ZINC000005399501, and AZ876 have the potential to penetrate the blood-brain barrier (Table 5). Lipophilicity or hydrophilicity are also important parameters of a molecule with a limited number of hydrogen bond donors and acceptors are more likely to achieve GI abroption because they can easily pass through the intestines’ cellular membrane and enter the bloodstream.33-35 Based on the calculated logP values all tested compounds proved to be lipophilic with consensus values ranging 3.6–4.98 (Table 6) There is less absorption of a medicine the more water soluble it is. All the selected candidates analysed water-solubility demonstrates moderately-soluble (Table 7).
Table 5: The basic physicochemical properties and computational descriptors of LXR-623, AZ876, and the newly identified molecules, as determined in SwissADME.
|
Features |
LXR-623 |
ZINC000005399501 |
AZ876 |
ZINC000021912941 |
|
Heavy atoms |
29 |
26 |
31 |
29 |
|
Aromatic heavy atoms |
21 |
12 |
12 |
12 |
|
Fraction Csp3 |
0.1 |
0.2 |
0.38 |
0.15 |
|
Rotatable bonds |
4 |
6 |
5 |
7 |
|
H-bond acceptors |
6 |
9 |
3 |
6 |
|
H-bond donors |
0 |
2 |
1 |
1 |
|
MR |
100.85 |
79.68 |
132.74 |
109.93 |
|
TPSA (Å2) |
17.82 |
74.78 |
78.1 |
118.23 |
Table 6: Lipophilicity of LXR-623, AZ876, and the newly identified molecule, determined in SwissADME.
|
Features |
LXR-623 |
ZINC000005399501 |
AZ876 |
ZINC000021912941 |
|
iLOGP |
3.53 |
1.92 |
3.7 |
2.83 |
|
XLOGP3 |
6.54 |
4.14 |
4.51 |
2.49 |
|
WLOGP |
8.69 |
7.08 |
4.56 |
7.52 |
|
MLOGP |
5.94 |
3.13 |
2.41 |
0.62 |
|
Silicos-IT Log P |
6.88 |
3.26 |
2.81 |
1.73 |
|
Consensus Log P |
6.32 |
3.91 |
3.6 |
4.04 |
Table 7: Water solubility prediction values of LXR-623, AZ876, and the newly identified molecule, determined in SwissADME.
|
Features |
LXR-623 |
ZINC000005399501 |
AZ876 |
ZINC000021912941 |
|
ESOL Log S |
-6.85 |
-4.87 |
-5.36 |
-3.82 |
|
ESOL Solubility (mg/ml) |
5.93E-05 |
5.39E-03 |
1.91E-03 |
6.24E-02 |
|
ESOL Solubility (mol/l) |
1.40E-07 |
1.35E-05 |
4.33E-06 |
1.51E-04 |
|
ESOL Class |
Poorly soluble |
Moderately soluble |
Moderately soluble |
Moderately Soluble |
|
Ali Log S |
-6.71 |
-5.42 |
-5.87 |
-4.62 |
|
Ali Solubility (mg/ml) |
8.21E-05 |
1.53E-03 |
5.91E-04 |
9.99E-03 |
|
Ali Solubility (mol/l) |
1.94E-07 |
3.82E-06 |
1.34E-06 |
2.41E-05 |
|
Ali Class |
Poorly soluble |
Moderately soluble |
Moderately soluble |
Moderately soluble |
|
Silicos-IT LogSw |
-9.49 |
-6.06 |
-6.74 |
-5.64 |
|
Silicos-IT Solubility (mg/ml) |
1.38E-07 |
3.48E-04 |
8.05E-05 |
9.43E-04 |
|
Silicos-IT Solubility (mol/l) |
3.26E-10 |
8.71E-07 |
1.83E-07 |
2.28E-06 |
|
Silicos-IT class |
Poorly soluble |
Poorly soluble |
Poorly soluble |
Moderately soluble |
Given the undeniable benefits of the oral route of administration, LXR-623 and ZINC000005399501 are expected to have low GI absorption, but AZ876 and ZINC000021912941 exhibit high GI absorption, which is a highly beneficial feature of a drug candidate. With very few exceptions, none of these substances are anticipated to function as CYP1A2 and CYP2D6 inhibitors, which are involved in the biotransformation of a number of significant medication classes. It is anticipated that all four of the substances under test will inhibit CYP3A4, CYP2C9, and CYP2C19. This could be detrimental because these CYPs are involved in the metabolism and excretion of numerous clinically prescribed medications. The target chemicals are not shown to be able to pass the BBB by the computational data.
The drug similarity, medicinal chemistry, and lead-likeness properties of the examined compounds were also computed. The characteristics of drug likelinesses are demonstrated by all of these compounds, with very few exceptions for Ghose, Egan, and Muegge breaches. The Abbot bioavailability score, which indicates the probability of a chemical exhibiting considerable Caco-2 permeability, was computed. Table 9 displays the data indicating that all examined compounds had a 0.55 likelihood of reaching the previously mentioned bioavailability for gastrointestinal absorption based on total charge, TPSA, and violation of the Lipinski. Synthetic accessibility of these compounds was within the range of 2.66-4.25 with no PAINS alerts and Brenk alerts (Table 9)
Table 8: Predicted pharmacokinetic parameters of the tested compounds.
|
Features |
LXR-623 |
ZINC000005399501 |
AZ876 |
ZINC000021912941 |
|
GI absorption |
Low |
Low |
High |
High |
|
BBB permeant |
No |
No |
No |
No |
|
Pgp substrate |
Yes |
No |
No |
No |
|
CYP1A2 inhibitor |
Yes |
No |
No |
No |
|
CYP2C19 inhibitor |
Yes |
Yes |
Yes |
Yes |
|
CYP2C9 inhibitor |
No |
Yes |
Yes |
Yes |
|
CYP2D6 inhibitor |
Yes |
No |
Yes |
No |
|
CYP3A4 inhibitor |
No |
Yes |
Yes |
Yes |
|
logKp (cm/s) |
‒4.24 |
‒5.8 |
‒5.78 |
‒7.06 |
Table 9: Drug likeness, medicinal chemistry, and lead-likeness parameters for the tested compounds.
|
Features |
LXR-623 |
ZINC000005399501 |
AZ876 |
ZINC000021912941 |
|
Lipinski violations |
1 |
0 |
0 |
0 |
|
Ghose violations |
1 |
1 |
1 |
0 |
|
Veber violations |
0 |
0 |
0 |
0 |
|
Egan violations |
1 |
1 |
0 |
0 |
|
Muegge violations |
1 |
0 |
0 |
0 |
|
Bioavailability Score |
0.55 |
0.55 |
0.55 |
0.55 |
|
PAINS alerts |
0 |
0 |
1 |
0 |
|
Brenk alerts |
0 |
0 |
0 |
0 |
|
Leadlikeness violations |
2 |
2 |
2 |
1 |
|
Synthetic Accessibility |
2.94 |
3.07 |
4.25 |
3.63 |
MD simulation of the known vs the newly selected compounds with LXR-α/β complexes
MD simulation was conducted to investigate the complexes of LXR-α-LXR-623, LXR-β-AZ876, and two novel ligand-proteins. Specifically, the RMSD of alpha carbon atoms, RMSF of all amino acid residues, and the number of hydrogen bonds of the selected drug-protein complexes were calculated for a duration of 50 ns. The RMSD of LXR-α-ZINC000005399501 and LXR-β-ZINC000021912941 showed stabilization after 10 ns of simulation, with average values of 1.95±0.20 Å and 1.85±0.33 Å, respectively (Table 10). However, the RMSD of LXR-α-LXR-623 became unstable after 45 ns of MD simulation, similar to the trend observed in the LXR-β-AZ876 complex (Figure 9). RMSF analysis, which explores the impact of drug molecule binding on the behavior of amino acid residues, revealed low RMSF values for LXR-α- ZINC000005399501 and LXR-β- ZINC000021912941, indicating reduced flexibility compared to the known protein-ligand complex. Additionally, the total number of hydrogen bonds between protein-drug complexes, which significantly contribute to the conformational stability of the complex, was calculated (Table 10). The LXR-α-ZINC000005399501 and LXR-β-ZINC000021912941 complexes exhibited more hydrogen bonds throughout the 50 ns simulation. Furthermore, binding free energy calculations were performed for each complex. The average binding free energy values were determined to be -28.33±0.39, -24.36±0.63, -15.21±0.06, and -17.89±0.35 kcal/mol for ZINC000005399501, ZINC000021912941, LXR-623 and AZ876, respectively.
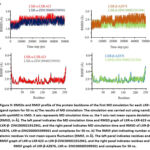 |
Figure 9: RMSDs and RMSF profile of the protein backbone of the first MD simulation for each LXR–ligand system for 50 ns |
Table 10: RMSD and binding free energy of the selected drugs–LXRs complexes, determined in QwikMD.
|
Complex |
RMSD (Å) |
Binding free energy (kcal/mol) |
|
LXR-α- ZINC000005399501 |
1.81±0.3 |
–23.33±0.39 |
|
LXR-β- ZINC000021912941 |
1.9±0.32 |
–22.36±0.63 |
|
LXR-α-LXR-623 |
2.49±0.71 |
–13.79±1.03 |
|
LXR-β-AZ876 |
2.24±0.41 |
–17.89±0.35 |
Discussion
Because of important regulator of glucose, fatty acid, and cholesterol homeostasis, LXRs are crucial for several major physiological processes including the metabolic system.10,50,51 Our in-silico study results of the distinct mRNA and protein expression of LXRα and LXRβ across the tissues also echoed their importance in physiological processes (Figure 1). There are milieu of reports stated the dysregulation of LXRs and the development of disease including atherosclerosis, Parkinsons disease, other metabolic disorders and cancer.10,12,52-55 These reports are aligned with our results of Gene disease association study, networks, and pathway enrichment analysis (Figure 2). LXR-α and LXR-β are linked to metabolic diseases like atherosclerosis and coronary heart disease, and are essential for physiological functions, interacting with enzymes, transcription factors, transporters, ion channels, and receptors (Figure 2).
Previous reports and our analysis of LXRs in physiological function and disease association intrigued us to find potential LXR modulators. Thus, we looked through the literatures and found out both endogenous and small molecule ligands targeting LXRs. Our search identified a list of LXR ligands and presented in table 1 and 2. Many of these ligands have been tested in preclinical phases.10,12,18,53-59 Only few of them are identified through screening or computer aided designing and tested in in-vitro settings.18,50,51,56,58 Therefore, our results of docking study as such binding affinities and presented interacting amino acids within 4 angstrom of the binding pocket would be important data addition to these known compounds (Table 1 and 2).
Our results of molecular docking, virtual screening, and pharmacokinetic in silico techniques indicated that LXR-623 and AZ876 had the best affinity with favorable drug-like features for LXR-α/β among these compounds (Figure 3, 4 and Table 5-9). For screening new ligands for LXR-α/β, we used a web-based server (http://www.swisssimilarity.ch/index.php). Swisssimilarity is integrated with many chemical structure computational models (e.g- FP2, ECFP4, MHFP6, Pharmacophore, ErG, Scaffold, Generic Scaffold, Electroshape, E3FP, etc.) and allows many chemical databases (e.g. ZINC, Drug Bank, ChemBL) for new ligand searching. In our study, we have identified two novel small molecules ZINC000095464663 and ZINC000021912925 from the ZINC drug-like database that exhibit the highest affinity for LXR, -12.3 and -11.7 Kcal/mol, respectively (Figure 6, 8 and Table 3, 4).
Basic physicochemical properties, ADME parameters, pharmacokinetic properties, druglike nature and medicinal chemistry friendliness of LXR-623, AZ876, andthe newly identified molecules are also favorable to the new drug molecule selection criteria for future drug development process (Table 5-9). Although, these estimations are acquired through the use of molecular descriptors derived from the molecules’ chemical structures and machine-learning methodologies. It is imperative to stress that, while in silico techniques for evaluating a drug’s ADME qualities can be useful in the early phases of drug development, in vivo investigations should always take precedence.37,41 Rather, these methods serve as a faster and more cost-effective means of investigating the pharmacokinetic characteristics of small molecules.41
Presently, the exploration and creation of novel drugs involves the use of computational technologies including MD simulations, virtual screening, pharmacokinetic investigations, and molecular docking.26-31,34-37,41 A deeper understanding of drug-protein interactions, the ability to improve the development and optimization of novel molecules, and the ability to predict the binding affinity between potentially active molecules and their target proteins through precise molecular simulation models and analyses are just a few of the major benefits of in-silico studies. These studies also offer cost and time savings compared to traditional wet lab experiments, as they allow for rapid evaluation of a large number of candidate molecules.38 The present study also leveraged these techniques to investigate the interaction with three LXR isoforms, leading to the identification of two compounds that hold promise for further laboratory studies on atherosclerosis. These compounds can be thoroughly evaluated for their safety and efficacy profile.
There are certain limitations on the types of evaluations conducted within this study of molecular docking, MD simulation, virtual screening and Chemico-pharmacokinetic strategies to identify novel compounds targeting LXRs.29 Molecular docking analysis has provided valuable insights into interactions between the target compounds and the LXRs; however, it remains essential to experimentally verify these findings. The results of this dry lab study may not always match the wet lab experimental validation technique’s predictions, and the procedure can be expensive and time-consuming. The proteins are highly dynamic in the biological environment and can experience significant conformational changes when bound by a ligand, molecular docking analyses concentrate on a single static interaction between the ligand and its target protein. Molecular docking analysis has been dependent on multiple approximations, including the exclusion of other proteins from the cellular milieu and the disregard of solvent impact.
The accuracy of the simulations may be compromised by these assumptions, which could lead to erroneous estimates. Given these constraints, there is potential to improve the study’s design by investigating new research directions using network pharmacology and molecular dynamics, which take into consideration the dynamic properties of proteins and possible interactions, the effects of solvents and ions, and the simulation of other cellular structures that might have an impact on the interaction.27,28,30
Conclusions
Both LXR-α and LXR-β exhibit distinct roles in the regulation of physiological cholesterol and lipids, as suggested by their distinct expression patterns, the associations between genes and diseases, and the enrichment of networks and pathways in both healthy and pathological tissues.
Through a comprehensive examination of the literature and a detailed analysis of molecular docking, simulation, and chemico-pharmacokinetic features, we narrated two new ligands (ZINC000095464663 and ZINC000021912925) with known agonists that have the ability to modulate LXRs. These findings may provide intuition for further investigation not only in-vitro but also in-vivo research for the future development of innovative therapeutic agents targeting LXRs.
Acknowledgment
We acknowledge the Department of Pharmacy, World University of Bangladesh, Dhaka for facilitating the project.
Conflicts of Interest
The authors do not have any conflict of interest
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article
Data Availability Statement
The datasets generated and/or analysed in this study are available from the corresponding author upon reasonable request.
Ethics approval
This research did not involve human participants, animal subjects, or any material that requires ethical approval
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required
Authors’ Contributions
Sarder Arifuzzaman conceived the study, interpreted the data and wrote the manuscript. Sarder Arifuzzaman revised the manuscript. Zubair Khalid Labu, Md. Harun-Or-Rashid, Farhina Rahman Laboni, Mst. Reshma Khatun, Md Sajib Ali, Shadek Hossain and Nargis Sultana Chowdhurywere involved in proofreading and revising the manuscript.All of the authors read and approved the final manuscript.
References
- Apfel R., Benbrook D., Lernhardt E., Ortiz M. A., Salbert G., Pfahl M. A novel orphan receptor specific for a subset of thyroid hormone-responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol Cell Biol. 1994;14(10):7025-35.
CrossRef - Liao Y., Wang J., Jaehnig E. J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):W199-W205.
CrossRef - Piñero J., Bravo À., Queralt-Rosinach N., Gutiérrez-Sacristán A., Deu-Pons J., Centeno E., García-García J., Sanz F., Furlong L. I. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017;45(D1):D833-D839.
CrossRef - Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Pontén F. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419.
CrossRef - Bunay J., Baron S., Lobaccaro J. A. LXRs are finally being adequately targeted in atherosclerosis. Ann Transl Med. 2018;6(Suppl 1):S28.
CrossRef - Repa J. J., Liang G., Ou J., Bashmakov Y., Lobaccaro J. M., Shimomura I., Shan B., Brown M. S., Goldstein J. L., Mangelsdorf D. J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14(22):2819-30.
CrossRef - Janowski B. A., Willy P. J., Devi T. R., Falck J. R., Mangelsdorf D. J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383(6602):728-31.
CrossRef - Joseph S. B., Castrillo A., Laffitte B. A., Mangelsdorf D. J., Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9(2):213-9.
CrossRef - Patel M. B., Oza N. A., Anand I. S., Deshpande S. S., Patel C. N. Liver x receptor: a novel therapeutic target. Indian J Pharm Sci. 2008;70(2):135-44.
CrossRef - Savla S. R., Prabhavalkar K. S., Bhatt L. K. Liver X receptor: a potential target in the treatment of atherosclerosis. Expert Opin Ther Targets. 2022;26(7):645-658.
CrossRef - Luo Y., Tan X., Zhang X., Li Y., Huang J., Deng Y. Effect of liver X receptor agonist TO901317 on cognitive function in APP/PS1 double transgenic mice with Alzheimer’s disease and the underlying mechanism. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2022;47(10):1324-1331.
- Mlera L., Offerdahl D. K., Dorward D. W., Carmody A., Chiramel A. I., Best S. M., Bloom M. E. The liver X receptor agonist LXR 623 restricts flavivirus replication. Emerg Microbes Infect. 2021;10(1):1378-1389.
CrossRef - Premaratne A., Ho C., Basu S., Khan A. F., Bawa-Khalfe T., Lin C. Y. Liver X Receptor Inverse Agonist GAC0001E5 Impedes Glutaminolysis and Disrupts Redox Homeostasis in Breast Cancer Cells. Biomolecules. 2023;13(2)
CrossRef - Yang M., Wang R., Sun J., Yu K., Chen B., Xu L., Zhao B., Wang H. The liver X receptor agonist TO901317 protects mice against cisplatin-induced kidney injury. Exp Biol Med (Maywood). 2015;240(12):1717-27.
CrossRef - Gao T., Qian T., Wang T., Su Y., Qiu H., Tang W., Xing Q., Wang L. T0901317, a liver X receptor agonist, ameliorates perinatal white matter injury induced by ischemia and hypoxia in neonatal rats. Neurosci Lett. 2023;793:136994.
CrossRef - Chuu C. P., Chen R. Y., Hiipakka R. A., Kokontis J. M., Warner K. V., Xiang J., Liao S. The liver X receptor agonist T0901317 acts as androgen receptor antagonist in human prostate cancer cells. Biochem Biophys Res Commun. 2007;357(2):341-6.
CrossRef - Koldamova R. P., Lefterov I. M., Staufenbiel M., Wolfe D., Huang S., Glorioso J. C., Walter M., Roth M. G., Lazo J. S. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J Biol Chem. 2005;280(6):4079-88.
CrossRef - Zhang W., Luo M., Zhou Y., Hu J., Li C., Liu K., Liu M., Zhu Y., Chen H., Zhang H. Liver X receptor agonist GW3965 protects against sepsis by promoting myeloid derived suppressor cells apoptosis in mice. Life Sci. 2021;276:119434.
CrossRef - Dianat-Moghadam H., Abbasspour-Ravasjani S., Hamishehkar H., Rahbarghazi R., Nouri M. LXR inhibitor SR9243-loaded immunoliposomes modulate lipid metabolism and stemness in colorectal cancer cells. Med Oncol. 2023;40(6):156.
CrossRef - Huang P., Kaluba B., Jiang X. L., Chang S., Tang X. F., Mao L. F., Zhang Z. P., Huang F. Z. Liver X Receptor Inverse Agonist SR9243 Suppresses Nonalcoholic Steatohepatitis Intrahepatic Inflammation and Fibrosis. Biomed Res Int. 2018;2018:8071093.
CrossRef - Jaye M. C., Krawiec J. A., Campobasso N., Smallwood A., Qiu C., Lu Q., Kerrigan J. J., De Los Frailes Alvaro M., Laffitte B., Liu W. S., Marino JP. Jr., Meyer C. R., Nichols J. A., Parks D. J., Perez P., Sarov-Blat L., Seepersaud S. D., Steplewski K. M., Thompson S. K., Wang P., Watson M. A., Webb C. L., Haigh D., Caravella J. A., Macphee C. H., Willson T. M., Collins J. L. Discovery of substituted maleimides as liver X receptor agonists and determination of a ligand-bound crystal structure. J Med Chem. 2005;48(17):5419-22.
CrossRef - NCT00379860. Study Evaluation LXR-623 in Healthy Adults. https://classic.clinicaltrials.gov/show/NCT00379860.
- NCT00613431. Effects of CS-8080 in Healthy Volunteers. https://classic.clinicaltrials.gov/show/NCT00613431.
- NCT00796575. Effects of Multiple Doses of CS-8080 in Healthy Volunteers. https://classic.clinicaltrials.gov/show/NCT00796575.
- NCT00836602. Multiple-Dose Study to Evaluate the Safety, Pharmacokinetics and Pharmacodynamics of BMS-779788 in Healthy Subjects. https://classic.clinicaltrials.gov/show/NCT00836602.
- Pinzi L., Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int J Mol Sci. 2019;20(18)
CrossRef - Sotriffer C. Docking of Covalent Ligands: Challenges and Approaches. Mol Inform. 2018;37(9-10):e1800062.
CrossRef - Ferreira L. G., Dos Santos R. N., Oliva G., Andricopulo A. D. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384-421.
CrossRef - Kaur T., Madgulkar A., Bhalekar M., Asgaonkar K. Molecular Docking in Formulation and Development. Curr Drug Discov Technol. 2019;16(1):30-39.
CrossRef - Meng X. Y., Zhang H. X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146-57.
CrossRef - Halimi M., Hajipasha A. Pharmacophore modelling, docking and molecular dynamic simulation studies in the discovery of potential human renin inhibitors. J Mol Graph Model. 2022;116:108272.
CrossRef - Arumugam R., Mani R., Venkatesan A., Sengamalai S., Natesan V., Kim S.-J. J. T. J. o. P. R. Molecular docking studies of natural compounds of naringin on enzymes involved in the urea cycle pathway in hyperammonemia. 2020;19(5):1037-1043.
CrossRef - Nirubama K., Narendhirakannan R., Rubalakshmi G., Vijayakumar N., Vinodhini M. J. K. R. J. Homology modeling and insilico approach of cleome gynandra-an indigenous medicinal plant. 2020;7(2):1-6.
CrossRef - Santos L. H. S., Ferreira R. S., Caffarena E. R. Integrating Molecular Docking and Molecular Dynamics Simulations. Methods Mol Biol. 2019;2053:13-34.
CrossRef - Bragina M. E., Daina A., Perez M. A. S., Michielin O., Zoete V. The SwissSimilarity 2021 Web Tool: Novel Chemical Libraries and Additional Methods for an Enhanced Ligand-Based Virtual Screening Experience. Int J Mol Sci. 2022;23(2)
CrossRef - Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.
CrossRef - Vamathevan J., Clark D., Czodrowski P., Dunham I., Ferran E., Lee G., Li B., Madabhushi A., Shah P., Spitzer M., Zhao S. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18(6):463-477.
CrossRef - Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3-26.
CrossRef - Uhlen M., Zhang C., Lee S., Sjöstedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F., Sanli K., von Feilitzen K., Oksvold P., Lundberg E., Hober S., Nilsson P., Mattsson J., Schwenk J. M., Brunnström H., Glimelius B., Sjöblom T., Edqvist P. H., Djureinovic D., Micke P., Lindskog C., Mardinoglu A., Ponten F. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352)
CrossRef - Fagerberg L., Hallström B. M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K., Asplund A., Sjöstedt E., Lundberg E., Szigyarto C. A., Skogs M., Takanen J. O., Berling H., Tegel H., Mulder J., Nilsson P., Schwenk J. M., Lindskog C., Danielsson F., Mardinoglu A., Sivertsson A., von Feilitzen K., Forsberg M., Zwahlen M., Olsson I., Navani S., Huss M., Nielsen J., Ponten F., Uhlén M. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397-406.
CrossRef - Potemkin V., Potemkin A., Grishina M. Internet Resources for Drug Discovery and Design. Curr Top Med Chem. 2018;18(22):1955-1975.
CrossRef - Ritter D., Goeritzer M., Thiele A., Blumrich A., Beyhoff N., Luettges K., Smeir E., Kasch J., Grune J., Müller O. J., Klopfleisch R., Jaeger C., Foryst-Ludwig A., Kintscher U. Liver X Receptor Agonist AZ876 Induces Beneficial Endogenous Cardiac Lipid Reprogramming and Protects Against Isoproterenol-Induced Cardiac Damage. J Am Heart Assoc. 2021;10(14):e019473.
CrossRef - WL D. The PyMOL molecular graphics system. 2002;
- Yuan S., Chan H. C. S., Filipek S., Vogel H. PyMOL and Inkscape Bridge the Data and the Data Visualization. Structure. 2016;24(12):2041-2042.
CrossRef - Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785-91.
CrossRef - Wu Q., Peng Z., Zhang Y., Yang J. COACH-D: improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018;46(W1):W438-W442.
CrossRef - Wass M. N., Kelley L. A., Sternberg M. J. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010;38(Web Server issue):W469-73.
CrossRef - Sterling T., Irwin J. J. ZINC 15–Ligand Discovery for Everyone. J Chem Inf Model. 2015;55(11):2324-37.
CrossRef - Dallakyan S., Olson A. J. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243-50.
CrossRef - Buñay J., Fouache A., Trousson A., de Joussineau C., Bouchareb E., Zhu Z., Kocer A., Morel L., Baron S., Lobaccaro J. A. Screening for liver X receptor modulators: Where are we and for what use? British journal of pharmacology. 2021;178(16):3277-3293.
CrossRef - She J., Gu T., Pang X., Liu Y., Tang L., Zhou X. Natural Products Targeting Liver X Receptors or Farnesoid X Receptor. Front Pharmacol. 2021;12:772435.
CrossRef - Zhao L., Lei W., Deng C., Wu Z., Sun M., Jin Z., Song Y., Yang Z., Jiang S., Shen M., Yang Y. The roles of liver X receptor α in inflammation and inflammation-associated diseases. Journal of cellular physiology. 2021;236(7):4807-4828.
CrossRef - Endo-Umeda K., Kim E., Thomas D. G., Liu W., Dou H., Yalcinkaya M., Abramowicz S., Xiao T., Antonson P., Gustafsson J. Å., Makishima M., Reilly M. P., Wang N., Tall A. R. Myeloid LXR (Liver X Receptor) Deficiency Induces Inflammatory Gene Expression in Foamy Macrophages and Accelerates Atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2022;42(6):719-731.
CrossRef - Han N., Yuan M., Yan L., Tang H. Emerging Insights into Liver X Receptor α in the Tumorigenesis and Therapeutics of Human Cancers. Biomolecules. 2023;13(8)
CrossRef - Alnaaim S. A., Al-Kuraishy H. M., Alexiou A., Papadakis M., Saad H. M., Batiha G. E. Role of Brain Liver X Receptor in Parkinson’s Disease: Hidden Treasure and Emerging Opportunities. Mol Neurobiol. 2024;61(1):341-357.
CrossRef - Li C., Chen H., Chen X., Li Y., Hua P., Wei J., Song C., Gu Q., Zhou H., Zhang J., Xu J. Discovery of tissue selective liver X receptor agonists for the treatment of atherosclerosis without causing hepatic lipogenesis. Eur J Med Chem. 2019;182:111647.
CrossRef - Ritter D., Goeritzer M., Thiele A., Blumrich A., Beyhoff N., Luettges K., Smeir E., Kasch J., Grune J., Müller O. J., Klopfleisch R., Jaeger C., Foryst-Ludwig A., Kintscher U. Liver X Receptor Agonist AZ876 Induces Beneficial Endogenous Cardiac Lipid Reprogramming and Protects Against Isoproterenol-Induced Cardiac Damage. J Am Heart Assoc. 2021;10(14):e019473.
CrossRef - Namba N., Noguchi-Yachide T., Matsumoto Y., Hashimoto Y., Fujii S. Design, synthesis and structure-activity relationship of 4-(1,1,1,3,3,3-hexafluoro-2-hydroxyisoprop-2-yl)phenylsilane derivatives as liver X receptor agonists. Bioorganic & medicinal chemistry. 2022;66:116792.
CrossRef - Gao T., Qian T., Wang T., Su Y., Qiu H., Tang W., Xing Q., Wang L. T0901317, a liver X receptor agonist, ameliorates perinatal white matter injury induced by ischemia and hypoxia in neonatal rats. Neurosci Lett. 2023;793:136994.
CrossRef

