
K. V. R. N. S. Ramesh¹, K. Ravishankar² and A. Bharathi³
¹Aditya Institute of Pharmaceutical Sciecnes.
²Sri Sai Aditya Institute of Pharmaceutical Sciences.
³Siddhartha College of Pharmaceutical Sciences, Surampalem - 533 437, East Godavari district India.
Corresponding Author E-mail:kantetiramesh@yahoo.com
Abstract
An in vivo pharmacokinetic study was performed in rabbits on prednisolone pure drug and a Sustained Relesase Formulation employing a more rapidly dissolving form of drug that was developed. A solution of prednisolone was used as reference standard. The products were given orally and at different time intervals blood samples were withdrawn and assayed for prednisolone content by reported HPLC method. From the plasma concentration time profiles the different pharmacokinetic parameters such as Cmax, Tmax, AUC, MRT, Kel, t1/2, Ka were calculated. While it was observed that in case of the pure drug the plasma concentrations (Cmax - 0.78 µg/ml ) were very low as also the AUC ( 7.6 µg-hr/ml), the solution form of the drug gave very high plasma concentrations of (Cmax - 4.15µg/ml ) and lower MRT values of 4.66 hrs , in the case of the sustained release formulations, the serum plasma concentrations (Cmax - 1.45 µg /ml; AUC – 20.67 µg-hr/ml ) were found to be intermediate between those obtained with solution form and the pure drug . Also the SR formulation gave serum concentrations that were stabilized and maintained over a narrow range and for longer periods. The relative bioavailability of prednisolone was found to be 92.25 % when compared to that of the solution taken as 100%.
Keywords
Pharmacokinetic; SR formulations
Download this article as:| Copy the following to cite this article: Ramesh K. V. R. N. S, Ravishankar K, Bharathi A. Pharmcokinetic Evaluation of Prednisolone SR Formulations Designed. Biomed Pharmacol J 2009;2(2) |
| Copy the following to cite this URL: Ramesh K. V. R. N. S, Ravishankar K, Bharathi A. Pharmcokinetic Evaluation of Prednisolone SR Formulations Designed. Biomed Pharmacol J 2009;2(2).Available from: http://biomedpharmajournal.org/?p=895 |
Introduction
Prednisolone is a corticosteroid anti-inflammatory, analgesic agent used in treatment of inflammatory diseases1. It is indicated for the treatment of primary or secondary adrenocortical insufficiency such as congenital adrenal hyperplasia and thyroiditis2. Prednisolone is a poorly water soluble drug. There are several reports 3-6 on the development of oral sustained release formulations of prednisolone.
The design of sustained release formulations of water in
soluble drugs is always difficult because in the absence of rapid dissolution of the drug, the release of the drug from the formulated product such as a matrix tablet is controlled more by the dissolution of the drug than by the nature of the polymer employed. It is common with oral sustained release formulations of poorly water soluble drugs that they show incomplete release from hydrophilic polymeric matrix systems. In a previously published research paper7, we reported the development of a sustained release matrix tablets of prednisolone. In that work, the dissolution rate of the drug was enhanced by solid dispersion and this dispersion was employed in the designing of matrix tablets to get faster release compared to that of matrix tablet which contained only the pure drug.
In this present study, the developed matrix tablet was subjected to in vivo pharmacokinetic evaluation in rabbits in comparison with that of pure drug and the drug solutionis employed as reference standard.
Materials and Methods
Materials
Prednisolone pure drug was obtained as a gift sample from M/s. Cipla Ltd. , Mumbai ; Ethyl acetate, Isoamyl alcohol, Hexane, Perchloric acid, Tetrahydrofuran and Water – all the chemicals are of HPLC grade.
The pharmacokinetic evaluation was done on the following products.
Prednisolone solution ( a solution of prednisolone in alcohol at 2 mg / ml concentration) Prednisolone pure drug Prednisolone SR formulation containing the more dissolving dispersion ( Pr : PVP at 1: 3 ratio )
The in vivo experiments were carried out in healthy rabbits as per the following experimental design and protocol. The protocol was approved by the IAEC.
Experimental Design
A crossover randomized block design ( RBD ) in which four rabbits each weighing 3.5 – 4.0 kg received one treatment ( product ) each every 15 days such that all the product are tested in all the four rabbits during the study. Thus, each treatment is replicated four times.
In Vivo Study Protocol
Rabbits weighing between 3.5-4.0 kg of either sex were used. They were fed on uniform diet. In the experiments, 18 hours fasted rabbits were used. The rabbits were not given food during the experiment, however they had free access to water.
After collecting the zero hour blood sample (blank), the product involved in the study was administered orally employing Ryle’s tube ( intubation tube ) at a dose equivalent to 2.0 mg of prednisolone. After administration 2 ml blood samples were collected from the marginal ear vein at 0.5, 1.0. 2.0, 3.0, 4.0 , 6.0, 8.0, 10.0 12.0, 18.0, and 24.0 hrs after administration of the product. The blood samples were centrifuged at 5,000 rpm and the serum separated was collected into dry tubes and stored under refrigerated conditions till assayed. Prednisolone content of the serum samples was determined by a known HPLC reported previously8. The serum prednisolone concentrations were calculated from a standard calibration graph. From the time versus serum concentrations data, various pharmacokinetic parameters such as peak concentration ( Cmax ), time at which peak concentration resulted ( Tmax ), area under curve ( AUC ), elimination rate constant ( Kel ), biological half life ( t1/2 ), percent absorbed to various times, the absorption rate constant ( Ka ), and mean residence time ( MRT ) were calculated in each case.
Estimation of Prednisolone in serum samples
Chromatographic Conditions
The chromatographic system consisted of a Model Agilent 1120 compact LC; samples were chromatographed at room temperature on a reversed phase C18 column ( Qualisil BDS CB, 150 X 4.6 mm, 5 µm ). The mobile phase consisting of (20: 80 v/v) tetrahydrofuran and water was used at a flow rate of 1.0 ml / min and the pressure was approximately 240 kg / sq.cm. Ultraviolet absorption was measured at 250 nm using Agilent variable wavelength detector.
Procedure
Prednisolone was isolated from 1.0 ml of human serum via precipitation with perchloric acid ( 60%), followed by liquid – liquid extraction with 5.0 ml of ethyl acetate: hexane : isoamyl alchol ( 80:19:1 v/v ). The samples were vortexed for 60 seconds and then centrifuged at 10,000 rpm for 10 minutes at room temperature. The supernant is then transferred to 16 mm x 150 mm test tune and mixed with 5.0 ml of ethyl acetate : hexane : isoamyl alcohol( 80:19:1 v/v ). After shaking samples for 45 to 60 seconds and centrifuging at 3200 rpm for 10 minutes, the organic layer was transferred to a clean 13 m x 100 mm test tube and evaporated to dryness at 40 o C . The samples were then reconstituted with the mobile phase and 20 µl was injected into the HPLC column.
Results and Discussion
The elimination rate constant Kel for prednisolone was found to be 0.29 hr -1 and the corresponding biological half life t1/2 was found to be 2.41 hrs. following the administration of prednisolone as a solution. The t1/2 value obtained in the present work is in good agreement with the earlier reported value of 2-4 hours 9 . The plasma concentration vs time profile obtained with different prednisolone products administered is shown in Fig.1 and the summary of the pharmacokinetic parameters is given in Table 1.
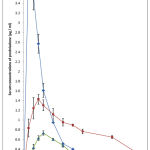 |
Figure 1 : Serum Concentrations of prednisolone following its oral administration as solution (♦), SR formulation (■) and as pure drug (▲).
|
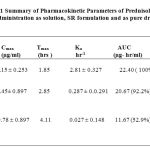 |
Table 1: Summary of Pharmacokinetic Parameters of Prednisolone after administration as solution, SR formulation and as pure drug. |
Application of Wagner – Nelson method to the serum concentrations data indicated a very rapid absorption of prednisolone given orally in solution form. The absorption rate constant Ka is found to be 2.81 ± 0.327 hr-1 and in one hour 87.67 ± 0.425 % was absorbed. Peak serum concentration of 4.15 ± 0.253 µg/ml was observed at 1 hour after administration and latter the concentrations declined rapidly.
Whereas when prednisolone was administered as pure drug there is found to be very low rate of absorption as evidenced by the low Ka value (0.027 ± 0.148 hr-1 ) and at the end of 4 hours, only 37 ± 0.661 % of drug was found to be absorbed. When prednisolone SR formulation was administered, serum concentrations were found to be intermediate between that of when solution form and the pure drug powder were given. For example, the peak prednisolone concentration was found to be 1.45± 0.897 µg/ml. Application of Wagner – Nelson method to the serum concentration data indicated slow absorption of prednisolone from the SR products compared to that from the solution but at the same time the absorption rate is found to be higher than that obtained with pure drug. From the SR formulation the absorption rate constant Ka is found to be 0.287 ± 0.0.291 hr-1 which is 10 fold lower than the absorption rate constant observed with the solution form but it is found to be higher by 10.6 fold when compared to that of pure drug. This goes to prove that the prednisolone bioavailability from the SR formulations employing the more dissolving solid dispersion form is higher that that of pure drug.
Additionally it is also evident that the prednisolone concentrations were stabilized and maintained over a narrow range and for longer periods of time in the case SR products. For example the serum concentrations were maintained in the range of 0.8 -1.0 µg/ml during the period 1-11 hrs with the SR formulations. The mean residence time ( MRT ) was found to increase from 4.66 ± 0.345 hrs for prednisolone solution to 9.64 ± 0.455 hrs with the SR formulation. SR formulation showed a relative bioavailability of 92.25% ( AUC – 20.67 µg-hr/ml ) when compared to prednisolone solution taken as 100% whereas when prednisolone pure drug was administered, the AUC value is found to be only 7.6 µg-hr/ml.
Conclusion
Thus the results of the pharmacokinetic studies carried out on the SR formulation of prednisolone developed by employing the solid dispersions of prednisolone indicated the drug was released and absorbed over longer periods of time which in turn maintained the serum concentrations within a narrow range for extended periods of time. Thus the present in vivo study corroborates the usefulness of the approach of employing solid dispersions of prednisolone in designing formulations ( as reported in our previous communications ) for sustained release of prednisolone.
Acknowledgement
The authors express their sincere gratitude to the management of Aditya Academy, Kakinada, for the facilities provided to carry out the research work
Refrencess
- Paul-Clark, M.J., Mancini, L. and Del Soldato P. “Potent antiarthritic properties of a glucocorticoid derivative, NCX-1015, in an esperimental model of arthritis”. Proc. Nat. Acad. Sci. USA, 99, 1677-1682, (2002)
- Drug Bank – Drug Bank Category Browser. URL: redpoll.pharmacy.ualberta.ca/drugbank/cat_browse.htm. (2006)
- Chen JH, and Shagufta M, “Preparation and characterization of prednisolone-poly (hydroxybutyrate-co-hydroxyvalerate) nanoparticles”. 37(6), 473-6, 2002
- Shinya, S, Teruko .I and Masaki O. (1994) “The Controlled Release of Prednisolone Using Alginate Gel.” Pharmaceutical Research, 11, 272 – 277, (1994)
- Rao, VM, Haslam J.L and Stella V.J. “ Controlled and complete release of a model poorly water-soluble drug, prednisolone, from hydroxypropyl methylcellulose matrix tablets using (SBE)(7m)-beta-cyclodextrin as a solubilizing agent”. J Pharm Sci.; 90(7), 807-16, (2001)
- Di Colo G,, Baggiani A, and Zambito Y, “ A new hydrogel for the extended and complete prednisolone release in the GI tract.” Int J Pharm., 310(1-2), 154-61. (2006.)
- Ramesh K.V.R.N.S., Ravishankar,K and Bharathi A. “Design and in vitro evaluation of controlled release matrix tablets of prednisolone,” Int.J.Chem.Sci., 5(3), 1053-1072, (2007)
- Scott Penzak, R and Elizabeth, T. “Prednisolone pharmacokinetics in the presence and absence o fritonavir after oral prednisolone administration to healthy volunteer” , J Acquir Immune Defic Syndr. ,40(5), 573-580.,(2005)
- Med Safe – Information for health care professionals URL :http://www.medsafe.govt.nz/profs/Datasheet/a/Apoprednisonetab.htm, (2007)

