
Bahareh Hashemi1, Azim Akbarzadeh2* and Mehdi Arjmand3
1Department of Chemical Engineering, Science and Research Branch, Islamic Azad University, Tehran, Iran 2Professor in Clinical Biochemistry, Pasteur Institute of Iran 3Associated professor in Department of Chemical Engineering, South Tehran Branch, Islamic Azad University, Tehran, Iran Corresponding Author Email: Azimakbarzadeh@pasteur.ac.ir
DOI : https://dx.doi.org/10.13005/bpj/929
Abstract
Today, nanotechnology has been imported in various fields of agriculture, pharmacy, medicine, electronics and food industries. Due to existence of the increasing need to develop safe, non-toxic and environmentally friendly methods (green nanotechnology), the wide range of nanostructures are made todays with the purpose of increasing the amount of healthy, non-toxicity and being environmentally friendly.Nanotechnology science and its products have found a good position in the pharmaceutical area, so that the global pharmaceutical market witnesses numerous pharmaceutical products using nanotechnology and many drugs such as drug delivering nanostructures are designed for less concentrated and more effective transferring of the drug and to reduce side effects as well as to enhance efficiency of treatment, that the chemotherapy drugs are some of them. So, the aim of this study was to provide formulation of nanoarchaeosomal paclitaxel and to evaluate parameters such as encapsulation, particle sizeand size distribution. To prepare archaeosome, the first phase was cultivation of Halobacterium salinarum and extraction of archaeosome from it. Then, it has been combined a certain proportion of cholesterol, archaeosome and paclitaxel with each other and the average diameter of the nanoarchaeosomal paclitaxel was measured with the aid of Zeta Sizer that was equal to 425 nm. The encapsulation efficiency of nanoarchaeosomal paclitaxel was obtained %96.32. Cell viability was examined by MTT assay on MCF-7 cell lines. Analysis on Cell viability percentage showed that dead effect of tumor cells in formulation of nanoarchaeosomal paclitaxel was more than free formulation. It means that the nanoarchaeosomal paclitaxel was more effective than paclitaxel in a freeform.
Keywords
Nanotechnology; Breast cancer; MCF-7 cell line; Archaeosome nano carrier; Paclitaxel
Download this article as:| Copy the following to cite this article: Hashemi B, Akbarzadeh A, Arjmand M. Providing the Formulation of Nanoarchaeosomal Paclitaxel and Evaluating Parameters of Encapsulation, Particle Size andSize Distribution. Biomed Pharmacol J 2016;9(1) |
| Copy the following to cite this URL: Hashemi B, Akbarzadeh A, Arjmand M. Providing the Formulation of Nanoarchaeosomal Paclitaxel and Evaluating Parameters of Encapsulation, Particle Size andSize Distribution. Biomed Pharmacol J 2016;9(1). Available from: http://biomedpharmajournal.org/?p=6911 |
Introduction
Cancer nanotechnology is the newest trend in treatment of cancer, this technology helps pharmacistto produce a pharmaceutical product with the highest therapeutic value and minimal side effects. Nanotechnology may be the only method that can be used for a unique function by creation of minimal side effects and damages to normal cells. Some of these methods that can be noted are systematic transfer systems, passive targeting, active targeting, intracellular transport, exposure to cell organelles and nanoparticle drugs (Smith et al, 2006).Studies on the use of nanotechnology in cancer treatment was performed in 2001 in Winship Cancer Institute. It was concluded during the studies that nanosized particles like as semiconductor quantum dots havestructural or magnetic or optical properties that are not observed in case of molecules or bulk solids and by binding the nanoparticles to tumortargeting ligands such as monoclonal antibodies and peptides or small molecules, these nanoparticles can be used with the high desire and expertise for targeted tumor antigens (biomarkers) and tumor blood vessels (Crook and Prestaydo, 1980). It was carried out extensive researches on the use of various nanoparticles in drug delivery during the years 2001 to 2004. So that in the last decades, nanoparticles have attracted much attention in researches as they are able to transfer drug. It has been started researches on drug-carrying particles or phospholipid vesicles known as liposomes. Conducted researches showed that liposome nano carrier scan increase effect level of the drug at a specified time and this makes the drug’s efficacy more than before and it would be diminished side effects and toxicityof drugs. Researches on nanoparticles that are used in the treatment of cancer as well as different drug delivery systems were expanding rapidly that the issue of drug delivery engineering was raised at the University of Louisiana in 2007.This was possible by engineering systems to be able to predictthe amount of drug delivery to the tumor at any time by obtainingthe efficacy time of drug on cancertumor,it can prevented the too much use of drugs. Thisresearch team created a huge development in the general fields of therapy and radiotherapy and immunotherapy and thermotherapy (Lacroix and Leclercq, 2004). In case of breast cancer, a man named Jan Park in San Francisco research center began doing experiments on drug delivery systems based on the nanoliposomes in 2005. He noted that the objective of drug delivery systems causes in fact an increase in the effectiveness and reduction of the toxicity in the reagents and anti-cancer drugs. Long cycle of carrier macromolecules and nano molecules such as liposome can cause the enhancement of permeability and increment in anti-cancer effect. Significant results of anticancer activities represented the effects of this drug delivery method by reducing the toxicity and an increase in retention time of many drugs such as Doxorubicin and Daunorubicin covered with liposome in the treatment of breast cancer in both monotherapy and combination therapy with chemotherapy (Salavati-niasar, 2009). The aim of this study was to provide formulation of nanoarchaeosomal paclitaxel and to evaluate parameters such as encapsulation, particle size and size distribution.
Materials And Method
Preparation of culture medium to grow Halobacterium salinarum
Halobacterium salinarumis a great halophilic, rod-shaped, gram-negativeand mandatory aerobic archaea that grows in areas with the temperature of 37 °C and pH of 7.2 and high concentrations of salt almost saturated. In this study, it was used the lineage of Halobacterium salinarum with specifications of IBR_M No: 10715 have bought from the genetic resources center. Methodwas in this way, the ingredients first were brought to the volumein the 500 ml flask with distilled water and its pH was set to the 7.2, the pH meter was used for that purpose. The pH of the solution was 6.7 at first and the basic solution was used to reach pH = 7.2. After setting pH, the solution was transferred into 5 250 ml flasks and the doors were closed and it was autoclaved in order to sterilize andit was used the flask label containing culture medium by name and date to paste autoclave’s special adhesive on the body of flasks. It was autoclavedunder conditions of 121 °C for 30 min. At the end of the autoclave, autoclave’s adhesive with diagonal lines got dark brown color which showedit was autoclaved properly. Culture medium that was prepared in this manner was the liquid culture medium for cultivation of archaeabacteria within the flask. Then, 10 ml of first stoke of Halobacterium salinarium bacteria was added to 5 flasks containing 200 ml culture mediumunder the hood.In order to better growthof bacteria and proper aeration, the flasks were putin incubator’s shaker device at 37 °C and speed of 170 rpm (for 10 days) and within 10 days, it would be determined the optical densitywith the spectrophotometer and it was drawn the logarithmic growth curve of Halobacterium.
The extraction of total polar lipids
After the growth of Halobacterium salinarum, the contents of the 5 flaskswere transferred into falconsthat autoclavedand measured previously, then were centrifuged (6000 rpm speed for 20 minutes) and the supernatant phase was taken out by Pasteur pipette and the precipitation offalconwasweighted and then the total polar lipids were extracted by means of blight and dyer method (Coleman and T songalis, 2002). Thus in the first stage,for each 30 gramsof the dry precipitation of bacteria, 420 ml of water was added to separate precipitation from the bottom of the falconand in order to the complete disintegration of bacteria,it was put within a beaker on stirrer for few hours. Then a separate solvent of chloroform, methanol and water with a ratio of 1: 2: 0.8 which was prepared was added and it was put on stirrer for 16 hours at 23 °C. It should be noted that the rotation speed of stirrer was not important at this stage. After 16 hours, the prepared suspension was transferred into falcon and was centrifuged by 4080g speed for 15 minutes at a temperature of 6 °C and the supernatant solution was collected. At this stage, the collected suspension was poured into the decantation funnel and total lipids were extracted by adding chloroform. By adding the required volume of chloroform, the solution got two-phased. The bottom phase which was the phase of chloroform should be removed and again with the addition of chloroform to the methanol and water phase, the entire lipids were extracted by repeating several times. The amount of required chloroform was the final volume of the supernatant solutions obtained after centrifugation from the previous stage divided by 3.8. To better understanding, for example: if a final volume of the collected supernatant solutions was 45 ml, chloroform should be added to the solution 4 times and each time 3 ml. After each level, the decantation funnel must be rotated like as number 8 for 10 minutes. Then for relaxing decantation funnel, it was placed quiet on the base for 10 minutes so that the separation phases becametotally visible. Then the bottom phase was removed. At this stage, the collected chloroform phase was put in a rotary evaporator at 50 °C and at 90 rpm to dry and discharge of chloroform, and after putting it in a rotary evaporator, the fine layer of lipids was made on the bottom of the flask that are the lipids dissolved in the chloroform.The fine lipid layer of the flask was washed with a very small amount of ethanol and more 4 times cold acetoneto precipitate totalpolar lipids extractions. And this solution was kept at -20 °C at least overnight, and then it was centrifuged at 7.00 rpm for 70 minutes. The supernatant solution was removed by Pasteur pipette and the white precipitationon the bottom of the falcon was total polar lipids extracted.
Preparation of nanoarchaeosome
Loading paclitaxel on archaeosome
The difference amount of empty falcon and falcon with white precipitation was the amount of total polar lipids extracted. The cholesterol was needed to apply nanoarcheozome on the total polar lipids extracted. Lipid production stages were done as above. Then it was added to the cholesterol solution (purchased from Sigma). Because the paclitaxel drug waslipophilic, it should be solved in solvents such as chloroform or methanol,so in this stage the drug was poured and the chloroform / methanol was added. Then the solution was put on the stirrer at 150 rpm at room temperature until completely dissolution. As the chloroform / methanol must be evaporated from the solution, so the solution was put again on a rotary evaporator, then PBS was added to the solution of the flask putting on the stirrer until completely dissolution.
Loading paclitaxel on PEG ylatedarchaeosome
At this stage, all was done same as the above. The difference was that the polyethylene glycol (PEG) was added besides cholesterol to the above composition. The drug was added after complete dissolution of PEG and cholesterol.
Preparation of nanoarchaeosome without paclitaxel
All of the above stages without paclitaxel must be repeated, thus the blank nanoarchaeosome was prepared.
Reducing the archaeosome’s size and production of nanoarchaeosome
After several hours of stirring on the magnetic stirrer and achieving a uniform solution to homogenize the vesicles, solutions were put into bath sonicator for 5 minutes in order to vesicles becoming homogeneous and same sized. Then solutions werehomogenized in 3 times in each, 1 minute with homogenizationdevise to reduce the particle size and then they were transferred into sterile glass vials puttingin the freezer.
Determination of particle size on the nano-scale
DLS or Zeta Sizer device
For this purpose, a certain amount of the two sonicatedsolutions of PEG ylatedarchaeosomal drug and archaeosome werediluted one to a hundred using physiologic serum (PBS) and then its absorbancewas read at a wavelength of 227 nm and the diameter of archaeosomes were measured in continue using Zeta Sizer devise (DLS) of Malvern company of England, the NanoZS model.
The standard curve
To draw a standard curve, 10 mg of paclitaxel drug must be dissolved in 10 ml of ethanol in which the initial concentration of the sample became 1 mg/ml. It was dissolved using vortex devise, then dilutedbycolddilution method. The optical density of solutions was measured using a spectrophotometer at a wavelength of absorbance of paclitaxel at 227 nm and then the standard curve was drawn.
Determination ofencapsulation efficiency and amount of drug loading
It was transferred the amount of 1 ml archaeosomal drug solution and the blank solution into two separate micro-tubes and they were centrifuged for 15 minutes at a speed of 18000 rpm to make clear the solution above the precipitation. Then the clear solution was placed in other micro-tubes. But if it was not clear, the colloidal state solution must be maintained in the refrigerator within 15 minutes to deposit due to the cold. Then it was again centrifuged, and their absorbance were measured at 227 nm. So according to the line equation which was the equation of standard curve, the unknown concentration of drugs would be achieved.
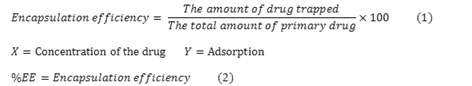
Cell cultivation
Cell culture room hasto be cleaned avoiding frommuchand unnecessary traffic and the door must be always closed. It is better to use the vacuum cleaners to clean the room. This room should not be closed to the microbiology sections or whereabouts of animals. Room doors should not be openedto the corridor that everyone comes and goes and the room windows should always be closed. This room should be empty of unnecessary equipment as much as possible to clean it easily. It must be used also stainless steel cabinets and tables as much as possible. The laminar hood is better to be stainless and its filters must be replaced at appropriate intervals. Washing tools of cell cultivation laboratory must be for the same room. The tables have to be cleansed from impuritieswith 70%alcohol each week. Each time after finishing the work of cell cultivation, inside the hoods should be wiped with 70% alcohol to prevent the growth of microorganisms in droplets sprayed from the culture medium. The incubator’s water should be changed every week and the copper sulfate 0.1% must beadded to it. Inside the incubator should be washed and sterilized every three months once with detergent and 70% alcohol.
Laminar hood
All steps of cell cultivation such as passage, culture medium preparation, culture mediumexchange, etc. must be done in a sterile environment. In cell cultivation laboratory, this environment is created by hoods equipped with special filters that are able to get particles larger than 0.2 microns. The air enters with pressure to the filter in these hoods and after getting its particles, the sterile air comes to the work environment. As a result, bacteria and spores suspended in the air causing pollution to the culture media may be ejected immediately from the workplace. The sterile air will enter into the hood containers horizontally or vertically. The hoods have ultraviolet light lamps that also help to sterilize the hood. The UV lamp should be off while working. Every day before starting work, the UV lamp of hood and culture room shall turn on for 30 minutes and then the surface of hood shall be cleansed with cotton and alcohol waiting a few minutesfor sterilizing the air inside the hood. The number of tools under the hood must be limited as much as possible not to block the air flow.
Carbon dioxide incubator
The incubator provides heat and moisture and CO2 needed for the growth of cells. Human cells need temperature of 37 °C for optimal growth. Incubator creates the unique and steady heat for growth of the cells by an automatic heat system and constant flow of air inside. Moreover incubator is also involved in setting the ********in culture media. Mostly usedculture media contain sodium bicarbonate. Bicarbonate in the culture medium providesthe appropriate buffer system to adjust pH of call culture media with the help of carbon dioxide entered into the incubator. The amount of carbon dioxide’s pressureis adjustable by a manometer connected to the CO2 gas cylinder and is often set at 5%. Usually, the pressure of CO2, the temperature and humidity of the incubator are shown by digital system and increase or decrease in each one makes device’s alarm active.To create humidity, the tray inside the set up should be filled with sterile water and copper sulfate is added to prevent contamination. Decrement of humidity can cause drying cell culture flasks and increment of humidity can increase the chance of contamination particularly with fungal spores.
Inverted microscope
Cultivated cellsshould be checked each day in case of contamination with fungi and bacteria. For this purpose, the inverted microscope equipped with phase control system was used. In addition to this microscope, normal optical microscope was needed for cell counting and determination of cell death.
Culture plates
Plastic or glass containers can be used. Plastic containers have reduced the risk of contamination of inadequate sterilization due to the disposabilityand reduce the washing and sterilization procedures. Cell culture flasks, centrifuge tubes, cryotubes and sampler are often plastic and disposable. Glass containers are often used for storing culture media.
Autoclave and Four (hot air oven)
All equipment used in cell cultivationshould be sterile. For this purpose, an autoclave is used for wet sterilization as well as Four for dry sterilization. Water and glass utensils, filters, etc., would be sterilized by autoclave and metal furniture and some glass equipment by the Four. To autoclave, the solution should be poured into a glass bottle and the door must be left open and covered with the aluminum sheet. It was autoclaved at 121 °C and at a pressure of 15 pounds per square inch for 30-60 minutes.After autoclave, the caps of bottlesshall immediately be fastened to prevent their contamination. Small objects such as filters can be wrapped in aluminum sheets and was autoclaved. After the autoclave in order to ensure, these devices can be dry sterilized again in Furnace.
Cell line
MCF-7cells were used in this study. This cell line is a breast cancer cell linewhich was isolated in 1970 by a 69-year old Caucasian woman. MCF-7 stands for Michigan Cancer Foundation 7, which refers to the institution in Detroit andHerbert Soule and his colleagues patented this cell linesin 1973.Michigan cancer charity now is known withthe name of Barbara Ann Karmanos Cancer Institute. Before MCF-7, researchers were unable to obtain cell lines ofbreast cancer that can survive more than a few weeks (Armstrong et al., 2000). MCF-7 cells are adherent cells; i.e.they must be attached to the bottom of the cell culture flaskfor the growth at first. Cell linewas used in this study was obtained from the cell bank of Iran Pasteur Institute in vials. This cell line is very sensitive and working with it requires great care. It is noteworthy that the first condition in cell cultivation is to provide sterile conditions. So before and after the work, work environmentand equipment should be sterilized.
Preparation of cell culture medium
The basis of all culturemedia are the same and include essential amino acids, vitamins, minerals and color indicators and the difference is indiversity or the amount of available amino acids and due to the cell varieties,each cell has its own culture medium. Culture medium of RPMI 1640 was prepared by Mooreet al. at Rosewell Park Memorial Institute in 1966. At first, thismedium was designed for the maintenance of linfoblastocells in the liquid media, however todays, it is used as the basic culture medium to feed a large group of adherent cells.RPMI 1640 is an economic medium for majority of cells and 60 to 70 percent of cell lines available have been compatible with this media (Kay et al, 2001). To prepare this culture medium, at first 1000 ml of deionized distilled water was poured into the glass caped container to autoclave, then the amount of 10.4 grams of MTT powder was dissolved in 1000 ml autoclaved deionized distilled water, and it was put on the magnetic stirrer for four hours and with the magnet disinfectedby 70% alcohol, so that it completely be solved with no visible particles in it.Approximately 15-30 minutes before filtering the culture medium, the two other materialsmust be added to the medium: two grams of sodium bicarbonate in 1000 ml, all culture media require bicarbonate because according to cultivation conditions at the temperature of 37 °C and 5%CO2, a water container is used in the incubator to prevent evaporation of water of culture medium and to produce the required humidity. So in this case, the CO2gas that is very soluble in water is dissolved in the culture medium and makes it acidic which is stressful to cells.So as to control the process of changing the pH, it is usedthe bicarbonate buffer (H, HCO3) as a tamponor a solution of seven and a half percent of bicarbonate.HEPES 99% was added in the two and a half grams in 1000 ml. HEPES isa buffer that prevents becoming the culture medium high acidic and basic and provides a suitable pH for cell growth that is the same as7.5. When using HEPES, it is better not to use the pH meter because it creates a lot of pollution and shall not be used. Filters can be used to sterilize the culture medium. The filtration basis is on pressure and vacuum. Here it was used the syringe filter with pore diameter of 0.22 micron and total diameter of 30 mm. The filters pores’ size was small and did not allow to entermycoplasma and fungi.These two organisms are the main pollutants of culture medium. To filter, the prepared culture medium was inserted into the 20 ml syringe, the syringe containing the culture medium was set on the filter and the sterilized filtered medium was discharged into sterilecapped and proper sized glasses. The vacuum pump creates pressure on the unfiltered solution with a vacuum which followed by a quick exit of solution from the filter into the sterile container. Finally, the filteredculture medium was maintained overnight at the temperature of 4 °C (in the refrigerator). In this case, if there was a bacteria, it grew and made the medium cloudy.
Cell cultivation and passage
Cells studied were cultivated in RPMI1640 medium containing 20.10% fetal bovine serum and were incubated at 37 °C containing 5% CO2 gas. Cell growth in culture medium requires a complex mix of nutrients as well as other compounds necessary for growth of cells. Culture medium has been composed of components with specific growth effects however, the FBS as an inexpensive affordable compound with essential components for proper cell growth may provide more suitable medium for the growth of cultivated cells. Serum is a part of the blood left that is formed after the coagulation and precipitation of its cellular components and consists of highly complex compounds of plasma proteins, growth factors, hormones, and so on. Most cell medium cultures involve about 10-20% FBS. Fetal bovine serum is stored in the freezer at -20 °C after purchasing. To use it, first it was put it in the refrigerator overnight. Then it was put at the 37 °C to be thawed completely. Then it was put at 56 °C for half an hour. This was due to remove possible contamination and disable serum complements. At the next step, it was pet at room temperature for 15 to 20 minutes to drop the temperature. Finally it was divided into 50 ml falcons to keepin the freezer, and every time a certain amount of it can be added to the culture medium. Since antibiotics can affect gene expression, it is better to reduce the possibility of contamination instead of using them in cell culture medium more carefully during the work. Bacterial contamination has separated the cells from the bottom and makes the cells medium cloudy, while hyphae is produced by fungal pollution and can be seen as fragments as colonies. Fungal contamination is very dangerous because samples containing fungus will contaminate all culture media in the incubator by producing spores. So as soon as seeing the fungus, the flask should be removed from the incubator. The cells produce acidic metabolites during the growth which causes a gradual color change from purple to yellow. For this reason, culture medium of cells usually was replaced every 24-48 hours. Pasteur pipette or sampler can be used to discharge previous culture medium. Given that cells release growth signals into the culture medium that a full discharge of them besides the culture medium brings the cells a lot of stress and pressure and the this stress could alter the pattern of gene expression, so while changing the culture media,10 to 20 percent of the medium was kept in flask. Then the fresh culture medium that was isotherm to the old medium was added to the flask containing the cells slowly from a side that did not suffer the cells adhered to the bottom and so the replacement was done.
The ways of cell passage
The basis: by the way of cell passage, cells are transferred from one place to another place to increase the number of cells. In laboratory conditions and when the appropriate growth conditions (37 °C, 5%CO2) in the presence of a suitable culture medium and the absence of pollution is provided for the cell, cells proliferate in the flask.When the entire surface of the substrate has been covered by the cells or had cell density over the capacity of substrate, cells must be transferred to another containers. This process is called cell passage. The passage is different depending on cells to be as suspense (suspended) or adherent. Suspense cells were transferred from the flask into falcon tubes and after centrifuging, the supernatant was discarded and the cell plate was transferred to the new falcon of culture medium. In the case of adherent cells, trypsin was used to separate the cells from the bottom of the flask.It should be noted that the temperature of trypsin should be at 37 °C when using. For this reason and to use it, it must be placed in a bain-marie after leaving out from the refrigerator. For cell passageat first,culture medium over the flasks must be completely discharged by Pasteur pipette or sampler, then two to three ml trypsin 0.25% was added to them and was remained for 3 to 5 minutes in this status in order to completely separation of cells from the bottom of the flask. Trypsin breaks cellular connections causing separation of the cells from the bottom of the flask and their floating. Care should be taken in the use of trypsin, if trypsin is took out earlier than the appropriate time,the number of cells is not separated and remains attached to the bottom and this will reduce the efficiency.If trypsin remains more than the proper time in the flask,it causes a change the shape of the cell surface proteins. As a result, cells can not adhere to the new flask. After appropriate time and to neutralize the trypsin effect, Supplied culture medium with serum culture medium was added to the flask and Pipetting slowly. The resulting suspension was transferred to falcon and centrifuged with 1000 rpm and for 7 minutes. After centrifugation, the supernatant was discarded and the cell plate with fresh culture medium was transferred to the new flask. It should be noted that the adherent cells after separation from bottom of the flask would be like as suspense and were easily transferable.According to the number of cells,it can be achieved at least two and up to four new cell flasks.
Freezing cells
The main goal of maintaining the cells in a frozen state is to preserve and access to sources of various cell lines without the need for continuous cultivation of them. Now several ways have been proposed to reduce damage to the cells during freezing. The most important thing to be successful in this process is slow freezing and rapid thawing of cells. In general, cells should be cooled quickly and be thawed within 3-5 minutes at 37 °C. Maintenance in liquid nitrogen has been the most successful method in keeping cultured cells. Cells used for freezing must have more than 90% viability and be absolutely free of kinds of microbial, viral contaminations, etc.
The ways of freezing cells
Steps offreezing the cells are like as processing of cell passage but all cells should be completely separated from the bottom of the flask with trypsin. Then, one to one and a half ml of solution named Cryosolution was added to cell precipitation resulting from the centrifugation. This solution contained 90% pure FBS and 10% DMSO. DMSO is a strong solvent acts as protective in the cell freezing process prevents formation of ice crystals and damage to the cells. DMSO is an excellent solvent, but it should be noted that is highly toxic to the cells and it must be careful to use it. The Cryosolution tubes were put at -20 °C for two hours and at -70 °C for 24 hours and then, available samples were transferred to the liquid nitrogen tank.This should be done in such a way that the cells be out of the freezer in the shortest possible time, because increase in the temperature of the vial causes damage to cells and decrease in living cells. The cells may be stored for a longer time by transferring them to liquid nitrogen tanks.
Thawing of thefrozen cells
To further cultivation(de-freezing) of frozen cells, cryo tubes containing frozen cells were transferred from nitrogen tank to the bain-marie at 37 °C.Precautionand use of safety equipment such as gloves and glasses was essentialwhen removing vials, because in case of not completely closing the vial’s tap,liquid nitrogen may penetrate into the vialand causes the explosion when thawing. After thawing, the contents of cryo tube have been transferred to falcon and the serum-free culture medium was added on it. As a result of this work, DMSO would be diluted and could not damage the cells. Then it centrifuged at 1000 rpmand for 7 minutes. At the next step, the serum-free culture medium was added again on the cell precipitation and centrifuged at 1000 rpm and for 7 minutes (same as previous step).Repeating of this action would lead to the complete disappearance of the effect of DMSO. Finally the cells with fresh culture medium containing 10-20% FBS were transferred to cell culture’s flasks. At this stage, the cells were spherical under the microscope, but recovered their original shape with the growth in the perfect medium. For example, if cells studied would be epithelial cells, they adhered to the bottom of the flask and get spindle after growth. In case of adherence of adherent cells to the bottom of the flask after 16 to 18 hours, the medium was replaced to exposure de-freezed cells which are very sensitive to fresh culture medium and FBS and to destroy the effect of remaining DMSO that supposed to be rarely.
The way of performingMTT assay
There are several methods to measure the proliferation and cell viability indirectly. Most of these methods measure the activity of a cellular enzyme in the living cells. MTT assay is one of the most known methods. MTT test isa laboratory and quantitative testfor measurement of cell proliferation in response to various agents and determination of the toxicity of these agents when they are affecting the cells. In fact, this technique helps to determine the toxicity of a particular drug. This technique was first used in 1983 by Mosmann. The basis of this method is on capability of mitochondrial reductase enzyme of living cells in reduction and conversion of yellow MTT tetrazolium rings to insoluble purple formazan crystals that are unable to cross the cell membrane.The crystals are then dissolved by the detergent to form solution. This detergent is usually dimethyl sulfoxide (DMSO) and sometimes SDS in dilute hydrochloric acid. Isopropanol is also an excellent solvent for formazan but it is usually used less due to the high cost.
During MTT assay, a reaction occurs in living cells as shown in below figure:
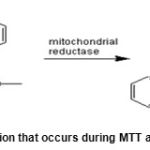 |
Figure 1: The reaction that occurs during MTT assay in living cells |
Optical density of the solution can be measured using spectrophotometer. Given that the above-mentioned reduction reaction occurs only in living cells which have active reductase enzyme, the numerical value obtained by this technique is directly linked to the number of living cells. In this measurement, the amount of produced formazan by cells treated with a special agent was compared with the formazan produced by control cells that did not receive special treatment and the amount of influence of that special factor was determined on cell death and cell growth inhibition by this comparison. As the amount of optical density read in this measurement would be less towards the control mode,it can be concluded that the number of living cells reduced and growth inhibitiontook place more.
The first day
Cells werecultivated in 96-house plate inMTT test, given that the houses of the plates were small and the cells cultivated on that houses could not be passaged during the test and to prevent excessive cell growth, the FBS percentage of the medium was decreased to five percent or two and a half percent which was typically 10%. In the first stage, cells were separated from the flask with trypsin. The process was similar to the cell passage but in the end, one ml of culture medium containing five percent FBS was added to cell precipitation instead of the culture medium containing 10% FBS and was Pipettin. It was added 10 microliter of this cell suspension the 10 microliter Trypan blue (this color inserts into membrane of dead cells but living cells do not absorb it.), then it was put on neubauer slide and the cells were counted. Cell counting done on the first day was crucial because it must be done in such a way that an equal number of cells insert into the houses. Then, the cell suspension was diluted so that there were 100 microliter culture medium containing 5% FBS and 10000 cells in each house.
The second day
Paclitaxel and nanoarchaeosome loaded with different concentrations (micro Molar) of paclitaxel were prepared in this day. Then the culture medium was discharged and instead, it was added 100 micrometers of adapted concentrations.
The fourth day
On the fourth day at first, 50 mg of MTT powder was dissolved in 10ml of PBS to provide the MTT solution with a concentration of mg/ml and the solution was diluted with PBS to 10-fold when used in staining to obtain the 0.5 mg/ml MTT solution. MTT powder can be stored up to 2 years at -20 °C, but it normally can be kept up to one month in the refrigerator.At the next stage, it was added 100 microliter of prepared solution to the each house. Plate was left for 3 hours in the incubator. Then it was gotten out from the incubator and the cells media were discharged and then to solve formazan produced, 100 microliter isopropanol was added to each house and plate was kept at room temperature for 30 minutes for formazan to dissolve well. At later stage, the optical density was measured using Elisa reader at 540 nm.
It was calculated the percentage of the survival in breast cancer cells with paclitaxel and nanoarchaeosomal paclitaxel according to the equations 3 and 4.
Determination of CI50 in paclitaxel and nanoarchaeosomal paclitaxel on MCF-7 cell lines
It was investigated all data (toxicity percentage) of the samples and control using Slacitistat, PCS-PHARM software and the exact amount of related CI50was determined in order to determine the dose of 50%fatality of toxic substances in breast cancer cell lines mentioned above
Results and Discussion
Evaluation the growth path of Halobacterium salinarum
The logarithmic growth standard curve of Halobacterium salinarum bacteria was drawn after several cultures of the bacteria and determined the amount of optical density at the wavelength of 600nm for 10 days. The results suggested that the mentioned bacteria had the highest growth rate on days 7 and 8. After this time, the bacteria had decreasing process, so the best time to harvest bacteria and lipid extraction was the same on day 8 (Figure 4).
 |
Figure 2: Spectrophotometer |
 |
Figure 3: Culturemedium containing Halobacterium Salinarum |
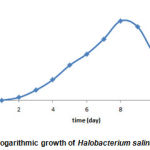 |
Figure 4: The logarithmic growth of Halobacterium salinarum bacteria |
Extraction of lipids
To extract lipids,the liquid of culture medium that the Halobacterium salinarum bacteria was growth in it was centrifugedin 6000 rpm and then the weight of dried biomass bacteria obtained that was almost 0.98 grams was stirred with a certain amount of distilled water for 2 hours on the stirrer and then this sample was brought to the volumeusing a solution made with ratios of 1: 2: 0.8 from chloroform / methanol / water and was put on the stirrer overnight in order to total lysis of the bacteria. After ensuring the lysis of bacteria (changing the color from red to yellow), the obtained solution was centrifuged. Then supernatant was carefully separated with Pasteur pipette so that the deposited material not to mix with it and it was transferred into the 100 ml decantation funnel and was washed 5 times each time with 3ml chloroform. Milky bottom phase (containing polar lipids) was separatedby opening the valve of decantation funnel and the sample obtained from decantation funnel was placed in rotary evaporation devise to form the fine lipid layer obtained by the lipid extract (Figure 5).
 |
Figure 5. The fine lipid layer derived from extraction |
Then, the fine lipid layer was washed with 5 ml of ethanol and more 4 times to its volume cold acetone was added and was kept at -20 °C for several hours. White deposits were collected from totalpolar lipids by centrifugation at 7100 rpmfor 10 minutes.
The standard curve of paclitaxel
The standard curve using the spectrophotometer
In accordance withthe table below, the optical density of the standard concentrations of paclitaxel solution was read by spectrophotometer at a wavelength 227nm (Table 1).
Table 1: The absorption of paclitaxel at 227 nm in different concentrations
| 0.818 | 0.446 | 0.259 | 0.168 | 0.146 | 0.125 | 0.112 | 0.116 | 0.099 | 0 | od |
| 0.0125 | 0.00625 | 0.00312 | 0.00157 | 0.00078 | 0.00039 | 0.000195 | 9.75E-05 | 4.88E-05 | 0 | con |
Based on these data, the standard curve of paclitaxel was drawn with excel software and the slope of the equation was found for next analysis (Figure 6).
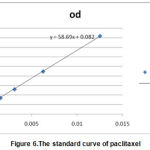 |
Figure 6.The standard curve of paclitaxel |
Determination of particle size in nanoscale and surface potential using Zeta Sizer (DLS)
In this study, sonicator and homogenizer devices were used to homogenize the formulation of nanoarchaeosomal paclitaxel and formulation of paclitaxel-free nanoarchaeosome.
 |
Figure 7: Sonicator |
 |
Figure 8: Homogenizer |
Then the size and surface charge of them were measured by Zeta Sizer and were compared with each other.
 |
Figure 9: Zeta Sizer |
 |
Figure 10: Size distribution curve of nanoarchaeo some without paclitaxel |
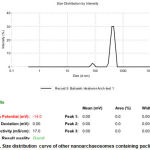 |
Figure 11: Size distribution curve of other nanoarchaeo somes containing paclitaxel
Click here to View figure |
Determinationof encapsulation efficiency of paclitaxel
Determination of encapsulation efficiencyusing high-speed centrifugation
It was possible to determine the concentration of paclitaxel which was not loaded on archaeosome using the equation derived from the standard curve and the amount of OD.
Y = 14.39X + .091
Optical density value of supernatant was equal to 0.795 that the encapsulation efficiencywas obtained according to the equation (2).
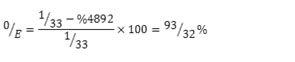
MTT results
MTT assay results was obtained as a plot by measuring optical density at the wavelength of 540 nm with Elisa Readerdevise based on the concentration of formulation of the used drug in comparison with the amount of cellular proliferation.MTT test was done in three turnsin accordance with the method described in last season. The viability of cells was determined after 48 h incubation. The viability rate of incubated cells with two types of drugs was calculated in proportion with the control wells. Results were stated as a percentage of viability of treated cells versus untreated cells and it can be calculated the CI50 of free and formulated drugs using the PHARM software. The average results of the three times are represented in the table below (Table 2).
Table 2. Average results of CI50on 7-MCF cell line
| Formulated drug | Free drug | Studied sample |
| 0.31 | 0.807 | CI50 |
It was compared the chart of CI50 for both compounds using pharm software.
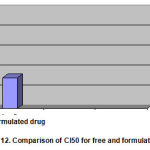 |
Figure 12: Comparison of CI50 for free and formulated drug |
Conclusion
The aim of this study was to provide formulation of nanoarchaeosomal paclitaxel and to evaluate parameters such as encapsulation, particle size and size distribution. In this study,it was observed that nanoarchaeosomal paclitaxel had fatality and damaging effect on MCF-7 cancer cells from the human breast tumor tissue in less daystowards free drug,so as a conclusion it can be said that regarding the benefits of nano-carriers for drug delivery applications,they can penetrate the cells through tiny capillariesdue to their small size resulting in efficient drug concentration in target locations and also stable and consistent drug release in this position for a period of several days or weeks.Encouraging results from liposomal drugs in the treatment and prevention of wide range of diseases in laboratory animals and humans shows that products based on nano-carriers will be produced for clinical applications in the future not far. Liposomes have the ability to cross biological barriers, protecting the drug against degradation, targeted drug delivery and release of the assigned treatment with an optimal dose. The great advantage of using nano-carriers towards release of drugs includes of protection of drugs from premature degradation and response to the biological environment, increase in the absorption of drugs into selective tissues through permeability and retention effect, control over pharmacokinetics and drug’stissue distribution profiles and improvement of intracellular penetration that are concerned as the therapeutic indices. However The major problem in using liposomal formulations is drug delivery to mucous channels, butother subject such as liquid nature of its preparation, proper viscosity and functional properties of liposomes can be earned by being incorporated in a suitable carrier, ithas proven that liposomes have relatively goodcompatibility with additive agents, the viscosity such as cellulose and polymers derived from acrylic acid (Carbopol). Despite many advantages of liposomes as drug delivery systems, instability and unsustainability of them are considered as one of the main problems associated with their use in pharmaceutical applications.Furthermore, it has been of interest the use of lipid nanoparticles called archaeosome that are a new type of liposomes made by the archaealpolar lipids which are concerned as a drug and vaccine delivery system. Lipids ofarchaeabacteria’s membranes have some special characteristics and chemical structure that make them different compared to conventional phospholipids such as phosphatidylcholine which are used in the production of liposome and they have some advantages such as ether bonds instead of ester bondswhich are more stable in a wide range of pH and also branched structure which reduces the permeability of the membrane and also saturated alkyl chains that cause high stability to oxidative damage as well as unusual spatial modes and stereo chemistry of main backbone that are resistant to the attack of enzymes like phospholipase. As a result archaeosomes with the highest stability are resistance to oxidative stresses, high temperatures and acidic or basic pH and performance of digestive enzymes. So they have been proposed as a substitute for conventional liposomes for more drug delivery or as new vaccine delivery systems. Existence of dams in body such as brain-blood dam, branching pathways in the lung and strong grafts of skin make it difficult to achieve the desired physiological targets for entrance of drugs that all these problems can be overcome with this technology. As mentioned, one of the most controversial cases in nano-therapy is to reduce or complete elimination of side effects.If active biological compounds influence on the target alone without the side effects, their beneficial effect will be significantly increased. In most cases of cancer treatment, drugs cannot diagnosebetween cells in normal divisionand neoplastic cells resulting in a problem in complete eradication of cancer and cause irreparable damages to healthy cells and tissues to the same time. It is known that perfect nano-therapy is achieved when the biological material can be combined with target’s specific nano-carrier system. Targeted delivery of biologically active compounds is the ability of concentrating biologically active substance in the favorable organ or tissue selectively and quantitatively as independent to the administered location.In recent study, the effect of archaeasomalpaclitaxel drug was investigated on MCF-7 cell lines of breast cancer. Archaeasomal technologyis a new approach to improve the pharmacokinetic properties of therapeutic proteins. Archaeosomes are attached as carriers with non-covalent protein, PEGylated liposome technology does not change the sequence of amino acids in a protein unlike methods such as mutagenicity, direct PGEylation, or fusion to carrier protein. In this research, some experiments were designed and conducted to measure the cytotoxicity of paclitaxel drug in free form and comparison of it with the archaeasomal form in laboratory conditions. At first, the Zeta Sizer (DLS) devise was used to determine the size of archaeasomes. It was reported that the average particle size of archaeasomalpaclitaxel was 425 nm and surface charge was-14 and an average particle size of the blank nanoarchaeo some without drug was 458.2 nm and so did for surface charge as -13. Zeta potential is a good indication of the repulsive value of interactions between colloidal particles which is closely linked with the stability of colloidal systems. If zeta potential of particles would be high positive or negative number, they will repel each other, they will have a stable suspension. The low zeta potential of particles, propulsive force will be small and particles can eventually dense resulting in suspension to become unstable.Under the conditions tested, zeta potential in both formulations was negative and previous studies have shown that negatively charged liposomes have high physical stability, small particle size and a relatively high percentage of drug trapping when compared to the positive charge. So by seeing the zeta potential in this study, it can be said that nanoarchaeosomalpaclitaxel had more physical stability than the blank nanoarchaeo some.The results of the drug loading calculated using supernatant separated from the solution showed that the amount of encapsulation of sample of archaeosome containing drug was ******** percent which was very convenient. The effect of archaeasomal paclitaxel and free drug samples was evaluated by MTT assay. MCF-7 cell line was used. The formulation of drug-free archaeosome had significantly less toxicity to the MCF-7 cell line. The results showed the nanoarchaeosomal drug had CI50 value less than the free paclitaxel.
References
- Salavati-niasarM. Nano-chemistry: manufacturing methods, evaluation of properties and applications. Sciense and knowledge publication;the first chapter (2009).
- Armstrong K, Eisen A, Weber B. Assessing the risk of breast cancer. N Engl J Med. 342 (8):564-71 (2000).
- Crook St, Prestaydo A, eds. Cancer and chemotherapy: introduction to neoplasia and antineoplastic chemotherapy. New York: Academic Press; pp.77-92 (1980).
- Coleman WB, Tsongalis GJ. The molecular basis of human cancer. Human press. Totowa, New Jersey; (2002).
- Key T, Verkasalo P, Banks E. Epidemiology of breast cancer. Lancet Oncol. 2: 133–140(2001).
- Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumors: an update. Breast Cancer Research and Treatment. 83 (3): 249-289(2004).
- Smith AM, Dave S, Nie S, True L, Gao X. Multicolor quantum dots for molecular diagnostics of cancer. Expert Rev MolDiagn. 6(2): 231–244 (2006).

