
Manuscript accepted on :February 06, 2016
Published online on: 28-02-2016
Plagiarism Check: Yes
Niloufar Akbarzadeh-Torbati* and Mehdi Shahraki
Department of Chemistry, University of Sistan and Baluchestan, P.O. Box 98135-674, Zahedan, Iran
DOI : https://dx.doi.org/10.13005/bpj/920
Abstract
The crystal structure of di-t-butyl-(N-2-indolinone-1-yl)-3- (triphenylphosphoranylidene) succinate has been determined by X-ray crystallography. This ylide was crystallized in the monoclinic system with space group P21/n and cell parameters a=15.0375(6) Å, b = 11.5130(5) Å, c = 19.1325(8) Å, β = 101.577(4)°, V = 3245.0(2) Å3, Dcalc= 1.273 g.cm-3 and Z = 4. The final R value is 0.0435 for 44084 reflections. This stable ylide existed as a mixture of two geometrical isomers in solution as a result of restricted rotation around the carbon-carbon partial double bond. The X-ray study showed that adjacent carbonyl group in the ylide moiety of compound had a resonance with bond of C=P and also, intermolecular C-H…O hydrogen bonds were effective on stability of the crystal structure. In addition, the stability of the two Z- and E- isomers and the most important geometrical parameters have been elucidated by natural population analysis (NPA) and atoms in molecules (AIM) methods.
Keywords
Ylide; X-ray Crystallography; Isomer; AIM; NBO
Download this article as:| Copy the following to cite this article: Akbarzadeh-Torbati N, Shahraki M. Assignment of the Major Geometrical Isomer of the Synthesized Stable Phosphorus Ylide: Crystallography and Theoretical Approaches. Biomed Pharmacol J 2016;9(1) |
| Copy the following to cite this URL: Akbarzadeh-Torbati N, Shahraki M. Assignment of the Major Geometrical Isomer of the Synthesized Stable Phosphorus Ylide: Crystallography and Theoretical Approaches. Biomed Pharmacol J 2015;9(1). Available from: http://biomedpharmajournal.org/?p=6508 |
Introduction
Organophosphorus compounds have emerged as important reagents and intermediates in organic synthesis [1]. An important group of this class is phosphorus ylides, which are used in many reactions and synthesis of organic compounds [2-8]. The prominent role of these compounds is to convert the carbonyl groups to carbon-carbon double bonds [9]. Michael addition of phosphorus (III) compounds such as triphenylphosphine to acetylenic esters leads to reactive 1, 3-dipolar intermediate betaines which are not detected even at low temperature [10]. These unstable species can be trapped by a protic reagent, ZH, such as methanol, amide, imide, etc. to produce various compounds e.g. ylides [11, 12]. In the present work, we reported the synthesis of the stable phosphorus ylides obtained by the reaction between triphenylphosphine and di-t-butyl acetylenedicarboxylate in the presence of indoline-2-one [8] (Fig. 1). These ylides usually exist as a mixture of the two geometrical isomers, although some ylides exhibit one geometrical isomer. Assignment of the two Z- and E- isomers are possible by some experimental methods such as 1H, 13C NMR, IR spectroscopy, mass spectrometry and elemental analysis data [13-16]. In order to gain a better understanding of the major isomer, the crystal structure of di-t-butyl-(N-2-indolinone-1-yl)-3-(triphenylphosphoranylidene) succinate was determined by X-ray crystallography. In addition, some theoretical methods have been performed for calculating the most important geometrical parameters and also relative energies of the both geometrical isomers.
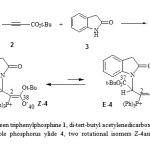 |
Figure 1: The reaction between triphenylphosphine 1, di-tert-butyl acetylenedicarboxylate 2 and indoline-2-one 3 for generation of stable phosphorus ylide 4, two rotational isomers Z-4and E-4 (major and minor, respectively) for ylide 4 |
Experimental Section
Material and Methods
Di-tert-butyl acetylenedicarboxylate, triphenylphosphine and indoline-2-on were purchased from Fluka (Buchs, Switzerland) and used without further purification. All extra pure solvents including dichloromethane, 1, 2-dichloroethane and THF obtained from Merck (Darmstadt, Germany).
Melting points and IR spectra was measured on an Electrothermal 9100 apparatus and a Shimadzu IR-470 spectrometer, respectively. The 1H, 13C, and 31P NMR spectra were recorded on a Bruker DRX-500 AVANCE instrument with CDCl3 as a solvent at 500.1, 125.8, and 202.4 MHz, respectively. The X-ray diffraction measurements of ylide 4 were made on an Oxford Diffraction Gemini diffractometer 100K (Mo-Kα radiation, graphite monochromator, λ = 0.71073 Å). The crystal of ylide 4 was solved by SHELXL-97 [17]. Absorption correction, data collection, cell refinement and data reduction have been carried out by using Oxford Diffraction Ltd [18-19], ORTEP (molecular graphics) [20], and WinGX (publication material) [21]. CCDC 760457 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Center.
Preparation of di-tert-butyl2-(N-2-indolinone-1-yl)3(triphenylphosphoranylidene) succinate 4
To a magnetically stirred solution of 2-indolinone (0.15 g, 1 mmol) and triphenylphosphine (0.26 g, 1 mmol) in 10 mL of ethyl acetate was added, dropwise, a mixture di-t-butyl acetylenedicarboxylate (0.23 g, 1 mmol) in 4 mL of ethyl acetate at -5oC over 10 min. After approximately 10 h stirring at room temperature, product was filtered and recrystallized from ethyl acetate.
Di-tert-butyl 2-(N-2-indolinone-1-yl)-3-(triphenylphosphoranylidene) succinate 4: White powder, yield 0.60 g (95%), m.p. 197-200°C, IR (KBr) (nmax, cm-1 ): 1736, 1710 and 1638 (C=O). Analysis: Calc: for C38H40NO5P (621.26) : C,73.4; H, 6.4; N, 2.25%; Found: C,73.3; H, 6.5; N, 2.1%. Major isomer (Z): 1H NMR (500.1 MHz, CDCl3): δH 0.97 and 1.57 (18H, 2s, 2OCMe3), 3.10 (1H, 2d, 2 JHH= 21.9 Hz, CH2), 5.47 (1H, d, 3JPH= 18.2 Hz, P=C-CH), 6.98 (1H, t, J=7.4 Hz, CHarom), 7.11 (1H, d, J=7.2 Hz, CHarom), 7.31 (1H, t, J=7.7 Hz, CHarom), 7.95 (1H, d, J=7.9 Hz, CHarom), 7.37 -7.67 (15 H, m, 3C6H5). 13C NMR (125.8 MHz, CDCl3): δC 28.2 and 28.4 (2CMe3), 35.45 (CH2), 37.7 (d, 1JPC =122.5 Hz, P=C), 54.5 (d, 2JPC= 16.2 Hz, CH), 77.3 and 80.6 (2OCMe3), 113.3-127.8 (6C, C6H4 ), 126.9 (d, 1JPC= 91.1 Hz, Cipso), 128.5 (d, 3JPC= 12.2 Hz, Cmeta), 131.9 (d, 4JPC= 1.9 Hz, Cpara), 133.7 (d, 2JPC= 9.6 Hz, Cortho), 168.7 (d, 3JPC= 11.8 Hz, C=O), 169.6 (d, 2JPC= 14.8 Hz, P-C=C), 173.7 (NC=O). 31P NMR (202.4 MHz, CDCl3): δP 22.5 (Ph3P +–C).
Results and Discussions
X-ray Crystallography
For establishing the major conformer, X-ray analysis of the ylide 4 is preceded. The single crystals were grown from a mixture of CHCl3/Et2O solution. A summary of the crystal data for ylide 4, experimental details and refinement results is given in Table 1. The molecular structure of ylide 4, showing the atom numbering scheme is illustrated in Fig. 2. Also, the molecular packing of ylide 4 is drawn in Fig. 3.
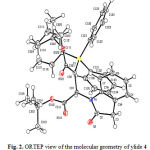 |
Figure 2: ORTEP view of the molecular geometry of ylide 4 |
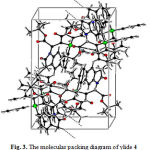 |
Figure 3: The molecular packing diagram of ylide 4 |
Table 1: Crystal data and structure refinement for C38H40NO5P 4
| C38H40NO5P 4 | Formula |
| 621.68 | Formula weight |
| 100(2) | Temperature(K) |
| 0.71073 | Wavelenght (Å) |
| Monoclinic | Crystal system |
| P21/n | Space group |
| 0.4×0.13×0.12 | Crystal size/mm3 |
| a = 15.0375(6) Å | Unit cell dimentions |
| b = 11.5130(5) Å | |
| c = 19.1325(8) Å | |
| β= 101.577(4)° | |
| 3245.0(2) Å3 | Volume |
| 4 | Z |
| 1.273 | Density(calculated) g cm-3 |
| 3.6161- 32.5621° | Theta ranges for data collection |
| 1320 | F(000) |
| 0.13 mm-1 | Absorption coefficient |
| -20 ≤ h ≤ 22 | Index ranges |
| -16 ≤ k ≤ 16 | |
| -25 ≤ l ≤ 28 | |
| 44084 | Data collected |
| 0.0772 | Unique data (Rint) |
| 412/ 0 | parameters / restraints |
| 0.0435, 0.071 | Final R1, wR2a (Obs. data) |
| 0.107, 0.0788 | Final R1, wR2a (all data) |
| 412 | Absolute structure parameter |
| 0.794 | Goodness of fit on F2 (S) |
| 0.393, -0.417 | Largest diff peak and hole /e.Å-3 |
| 760457 | CCDC |
aR1 = Σ||Fo|-|Fc||/Σ|Fo|, wR2 = [Σ[w(Fo2-Fc2)2]/Σ[w(Fo2)2
The X-ray study of ylide 4 shoewd some weak intermolecular C-H…O hydrogen bonds (Table 2) which are effective on stability of the crystal structure. Selected bond lengths, bond angles, and torsion angles of C38H40NO5P 4 are listed in Table 3. As can be seen in Tables 2 and 3, there is a good agreement between the obtained results emerged from these methods for crystal structure of ylide 4, E-4 (hypothetical) and Z-4.
Table 2: Hydrogen bond geometries of C38H40NO5P in the crystal packing (Å). Symmetry codes: (i) 1-X,1-Y,1-Z, (ii) 1/2-X,-1/2+Y,3/2-Z
| D -H…A | D -H | H…A | D…A | D – H…A |
| C3─H3A…O2i | 0.99a(1.09)b | 2.47a(2.74)b | 3.3535a(17)(2.4236)b | 149a (60)b |
| C31─H31…O2 | 1.00(1.08) | 2.42(2.34) | 2.7890(15)(2.7715) | 101(102) |
| C313─H31C…O311 | 0.98(1.08) | 2.45(2.46) | 3.0131(17)(3.0304) | 116(111) |
| C314─H31D…O311 | 0.98(1.09) | 2.50(2.48) | 3.0938(16)(3.0312) | 118(110) |
| C323─H32A…O321 | 0.98(1.07) | 2.43(2.42) | 3.0098(18)(2.9970) | 118(112) |
| C324─H32F…O321 | 0.98(1.08) | 2.48(2.42) | 3.0407(17)(2.9904) | 116(111) |
| C114─H114…O2ii | 0.95(1.07) | 2.39(2.49) | 3.2702(17)(3.1807) | 154(121) |
| C116─H116…O312 | 0.95(1.07) | 2.55(2.46) | 3.4452(16)(3.5107) | 156(164) |
a: The experimental data obtained from X-ray.
b: The theoretical data for the Z-isomer obtained at HF/6-31G(d,p) level.
On the basis of X-ray crystallography data, it is obvious that adjacent carbonyl group in the ylide moiety of ylide 4 (C321=O321, Fig. 2) has a resonance with bond of C32=P1. In the present work, single crystal X-ray diffraction of stable phosphorus ylide 4 was prepared from a solution involving 95% of major isomer (Z isomer, in accordance with 1H-NMR data). Herein X-ray analysis exhibited the Z-structure for this compound, the tert-butoxy and triphenylphosphine groups are placed in the same side of carbon-carbon double bond (Fig. 1.). On the other hand, the structural assignments of the Z-isomer as a major form have previously been reported for other phosphorus ylides [22, 15]. Herein the 1H NMR spectrum of ylide 4 exhibited two singlets at 0.97 and 1.57 ppm arising from the two tert-butoxy groups; this only occurs for the Z-isomer of ylide 4 because of the tert-butoxy group (at 0.97 ppm) is shielded due to the anisotropic effect of a phenyl group of triphenylphosphine. This phenomenon for the two tert-butoxy groups in the E-isomer could not be observed in the 1H NMR spectroscopy and chemical shifts for the two tert-butoxy groups in this isomer should be appeared near to each other, this imply that on other demonstration for confirmation of the Z-isomer in comparison with the E-isomer. On the basis of this information the results of previous works [8] should be corrected, and the major form should be reported for the Z-isomer of ylide 4 or other ylides.
Table 3: The selected Bond Lengths (Å), Bond Angles (°) and Torsion Angles for C38H40NO5
| (°) | Torsion Angles | (°) | Bond Angles | (Å) | Bond Lengths | ||
| 14.7a(2) [5.24]b |
C(31) C(32) C(321) O(321) |
110.15a(10) [112.95]b [116.60]c |
O(322) – C(321) – C(32) |
1.2295a(16) [1.2060]b [1.2180]c |
O(321) – C(321) |
||
| 179.62(11) [168.13] |
P(1) C(32) C(321) O(321) |
122.98(11) [122.04] [121.81] |
O(321) – C(321) – O(322) |
1.3721(16) [1.3466] [1.3341] |
O(322) – C(321) |
||
| -0.45(14) [-12.96] |
P(1) C(32) C(321) O(322) |
126.87(12) [125.00] [121.60] |
O(321) – C(321) – C(32) |
1.7208(13) [1.7268] [1.7386] |
P(1) – C(32) |
||
| -165.33(10) [-173.64] |
C(31) C(32) C(321) O(322) |
117.35(9) [119.93] [111.71] |
P(1) – C(32) – C(321) |
1.4179(17) [1.4292] [1.4257] |
C(32) – C(321) |
||
| -163.78(10) [-162.35] |
N(1) C(31) C(311) O(312) |
120.99(9) [121.31] [121.03] |
P(1) – C(32) – C(31) |
1.5094(18) [1.5075] [1.5036] |
C(31) – C(32) |
||
| -105.00(13) [-72.13] |
N(1) C(31) C(32) C(321) |
119.86(11) [118.42] [127.15] |
C(31) – C(32) – C(321) |
1.4687(15) [1.4684] [1.4697] |
N(1) – C(31) |
||
| 24.16(16) [38.50] |
C(311) C(31) C(32) C(321) |
110.12(10) [112.63] [112.01]c |
N(1) – C(31) – C(311) |
1.5315(18) [1.5340] [1.5354] |
C(31) – C(311) |
||
| 90.68(12) [124.65] |
N(1) C(31) C(32) P(1) |
125.80(12) [123.66] [124.55]c |
O(311) – C(311) – C(31) |
1.1992(16) [1.1904] [1.1896] |
O(311) – C(311) |
||
| -140.16(9) [-101.16] |
C(311) C(31) C(32) P(1) |
107.27(10) [111.10] [110.27]c |
O(312) – C(311) – C(31) |
1.3463(15) [1.3175] [1.3196] |
O(312) – C(311) |
||
| 63.82(13) [174.18] |
C(32) C(31) C(311) O(312) |
||||||
|
-120.58(14) [116.52] |
C(32) C(31) C(311) O(311) |
||||||
| a: The experimental data obtained from X-ray for ylide 4 compound.
b: The theoretical data for the Z-4 isomer obtained at HF/6-31G(d,p) level. c: The theoretical data for the hypothetical E-4 isomer obtained at HF/6-31G(d,p) level.
|
|||||||
Theoretical studies
For assignment of the two Z and E isomers as a minor or major form in phosphorus ylide 4 containing an indoline-2-one, first the Z- and the E- isomers were optimized for all ylide structures at HF/6-31G(d,p) level of theory [14, 23] by Gaussian 03 package program [24]. The relative stabilization energies in both the geometrical isomers have been calculated at HF/6-31G (d,p) and B3LYP/6-311++G** levels. Atoms in molecules (AIM) [25], natural population analysis (NPA) methods and CHelpG keyword at HF/6-31G(d,p) level of theory have been employed in order to gain a better understanding of most geometrical parameters of both the E-4 and the Z-4 of phosphorus ylides. The numbers of critical points and intramolecular hydrogen bonds as well as the charge of atoms that constructed on the Z- and E- isomers have been recognized. In addition, Jx-y, the values of proton and carbon coupling constants and also chemical shifts (δHiso, δCiso) have been calculated at mentioned level using SPINSPIN keyword. The relative stabilization energies for the two isomers (Fig. 4) are compared and it is concluded that Z-4 isomer is more stable than E-4 form (2.55 kJ/mol) at HF level.
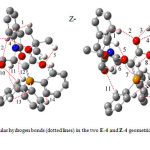 |
Fig. 4: Intramolecular hydrogen bonds (dotted lines) in the two E-4 and Z-4 geometrical isomers of stable ylide 4 |
Further investigation was undertaken in order to determine more effective factors on stability of the two Z- and E- isomers on the basis of AIM calculations at HF/6-31G (d,p) level of theory by the AIM2000 program package [26]. In recent years, AIM theory has often applied in the analysis of H-bonds. In this theory, the topological properties of the electron density distribution are derived from the gradient vector field of the electron density Ñr(r) and on the Laplacian of the electron density Ñ2r(r). The Laplacian of the electron density, Ñ2r(r), identifies regions of space wherein the electronic charge is locally depleted [Ñ2r(r) > 0] or built up [Ñ2r(r) < 0] [27]. Two interacting atoms in a molecule form a critical point in the electron density, where Ñr (r) = 0, called the bond critical point (BCP). The values of the charge density and its Laplacian at these critical points give useful information regarding the strength of the H-bonds [28]. The ranges of r(r) and Ñ2r(r) are 0.002 – 0.035 e/a03 and 0.024 – 0.139 e/a05, respectively, if H-bonds exist [29]. The AIM calculation indicates intramolecular hydrogen bond critical points (H-BCP) for the two Z-4 and E-4 isomers. Intramolecular H-BCPs along with a part of molecular graphs for the two rotational isomers are shown in Fig. 4 (dotted line). Most important geometrical parameters involving some H-bonds (bond length and their relevant bond angle) are reported in Table 4. The electron density (r)×103, Laplacian of electron density Ñ2 r(r)×103, and energy density -H(r)×104 are also reported in Table 5.
Table 4: Most important rotation parameters corresponding to H-bonds (bond lengths and their relevant bond angles) for the two E- and Z- isomers in ylide 4. Bond lengths in angstroms and bond angles in
degrees
| H-Bonding | E-4 | Z-4 |
| C82-H85…O58 | 2.18a | 2.47a |
| (114.3)b | (111.3)b | |
| C4-H7…O58 | 2.53 | 2.46 |
| -164.8 | -164.2 | |
| C26-H29…O56 | 2.46 | 2.34 |
| -128.6 | -120.6 | |
| C78-H80…O58 | 2.32 | 2.48 |
| -102.1 | -110.3 |
a Bond lengths on the basis of theoretical calculation
b Bond angles on the basis of theoretical calculation
Table 5. The values of px103,∇2p2x103 and Hamiltonian –H(r) x104 for the two Z-4 and E-4 isomers of ylide 4 calculated at the hydrogen bond critical Points. All quantities are in atomic units
| E | p x 103 | ∇2px103 | H(r) x104 | Z | r ´103 | ∇2p x103 |
H(r) x104 |
| 1 | 6.192 | 20.50 | -10.11 | 1 | 3.441 | 11.68 | -6.857 |
| 2 | 3.754 | 12.69 | -7.236 | 2 | 13.03 | 52.77 | -16.11 |
| 3 | 7.593 | 34.52 | -20.30 | 3 | 13.12 | 49.71 | -11.88 |
| 4 | 6.316 | 25.63 | -10.77 | 4 | 13.09 | 49.85 | -12.31 |
| 5 | 14.25 | 54.31 | -12.45 | 5 | 11.84 | 45.87 | -12.86 |
| 6 | 14.96 | 56.92 | -12.63 | 6 | 11.60 | 45.20 | -13.22 |
| 7 | 11.26 | 43.46 | -11.01 | 7 | 8.985 | 32.38 | -6.484 |
| 8 | 11.64 | 45.15 | -12.84 | 8 | 8.047 | 33.12 | -12.21 |
| 9 | 11.40 | 44.62 | -13.40 | 9 | 2.644 | 8.139 | -4.577 |
| 10 | 7.234 | 27.32 | -8.087 | 10 | 2.790 | 8.974 | -4.899 |
| 11 | 12.20 | 43.84 | -16.42 | 11 | 9.166 | 36.60 | -12.41 |
| 12 | 9.653 | 37.96 | -11.53 | 12 | 9.543 | 34.40 | -16.45 |
| 13 | 9.918 | 36.09 | -16.97 | 13 | 9.518 | 32.86 | -14.50 |
| 14 | 9.191 | 31.61 | -14.71 |
A negative total energy density at the BCP reflects a dominance of potential energy density, which is the consequence of accumulated stabilizing electronic charge [30]. Herein, the number of hydrogen bonds in E-4 and Z-4 are 8 and 7, respectively. The values of (r and Ñ2r(r) )×103 for those are in the ranges 0.028-0.042 and 0.031-0.051 e/a05, and 0.012-0.056 and 0.08-0.052 e/a05, respectively. In addition, the Hamiltonian [-H(r) ×104] are in the ranges 8.08-20.3 and 4.5-16.11 au, respectively (Table 6). These HBs show Ñ2r(r) > 0 and H(r) < 0, which according to classification of Rozas et al. [29] are medium-strength hydrogen bonds. In ylide 4 the dipole moment for the E-4 isomer (3.17 D) is smaller than the Z-4 isomer (4.35 D), and the value of -Htot (=å-H(r) ×104) for the Z-4 isomer (144.68 au) is smaller than the E-4 isomer (163.80 au). Although, dipole moment, number of hydrogen bonds and -Htot in both of the E-4, taken altogether, appear as two factors on stability of the E-4, but these values are not able to provide a power full effect as a dominance of potential energy. It seems that other factors i.e. other intramolecular exception of H-BCPS and anisotropic effect of phenyl group in ylide 4 containing triphenylphosphine within the structure of the Z-4 isomer could be accounted against mentioned factors for changing the order of relative energy between the Z- and E- isomers. As a result, the Z-4 has a considerable stability with respect to the E-4 (Table 4). As expected, according to theoretical calculations the relative energy at HF/6-31G (d,p) level, are compatible with experimental results from the X-ray crystallography, 1H , 13C and 13P NMR spectroscopy which indicate the Z-4 is more abundance isomer (Table 4).
Table 6: The most important geometrical parameters involving the value of -Htot/au, dipole moment/D and the number of hydrogen bonds for the two Z- and E- isomers of ylide 4
| Geometrical isomer | -Htot/au | dipole moment/D | number of hydrogen bond |
| E-4 | 171.2 | 3.17 | 14 |
| Z-4 | 144.8 | 4.35 | 13 |
Table 7: The charges on different atoms for both Z- and E- isomers in ylide 4 calculated at HF/6-31G(d,p) level
|
Number of atom |
Z-4a |
E-4a | Z-4c |
E-4c |
| P1 | 3.25 a
(3.22´1-1)b (1.87)c |
3.24
(2.65´1-1) (1.87) |
3.24
(3.81´1-1) (1.87) |
3.25
(2.58´1-1) (1.87) |
| C2 | -7.63´1-1
(-7.06´1-1) (-8.93´1-1) |
-7.81´1-1
(-6.82´1-1) (-8.80´1-1) |
-7.37´1–
(-7.49´1-1) (-8.90´1-1) |
-7.6´1-1
(-7.79´1-1) (-8.69´1-1) |
| C36 | 7.48´1-1
(4.83´1-1) (-1.06´1-1) |
7.48´1-1
(4.49´1-1) (-1.03´1-1) |
7.52´1-1
(5.17´1-1) (-9.91´1-2) |
7.66´1-1
(6.09´1-1) (-1.03´1-1) |
| C38 | 1.85
(9.11´1-1) (9.68´1-1) |
1.85
(9.07´1-1) (9.63´1-1) |
1.85
(9.25´1-1) (9.76´1-1) |
1.86
(1.02) (9.68´1-1) |
| O39 | -1.40
(-7.09´1-1) (-7.79´1-1) |
-1.40
(-7.14´1-1) (-8.03´1-1) |
-1.40
(-7.16´1-1) (-7.90´1-1) |
-1.42
(-7.12´1-1) (-8.18´1-1) |
| O40 | -1.28
(-4.54´1-1) (-6.68´1-1) |
-1.27
(-4.13´1-1) (-6.52´1-1) |
-1.31
(-5.77´1-1) (-6.93´1-1) |
-1.27
(-7.31´1-1) (-6.70´1-1) |
| a) Calculated by AIM method.
b) Calculated by CHelpG keyword. c) Calculated by NPA method. |
||||
Also, the charge on different atoms which are calculated by AIM and NPA methods and also CHelpG keyword at HF/6-31G(d,p) level are reported in Table 7 for the two Z- and E- isomers of ylide 4. There is a good agreement between the results in three methods. The differences between the charge on P1 atom in relation to Z-4 and E-4 are very close to each other in three methods.
Furthermore the individual chemical shifts have been characterized by NMR calculations at the mentioned level for the E-4 and Z-4 geometrical isomers. The total spin–spin coupling constant is the sum of two isomers. The paramagnetic spin-orbit (PSO) diamagnetic spin-orbit (DSO) Fermi-contact (FC) and spin-dipole (SD) terms and the value of chemical shifts (d) and coupling constants (Jx-y) are reported in Tables 8 and 9. As can be seen there is a good agreement between both the experimental [8] and theoretical chemical shifts (d) and coupling constants (Jx-y). In the present work molecular structures of ylide 4 involving three large atoms such as sulfur, phosphorus and nitrogen are huge with the large numbers of other atoms, for this reason, employment of basis set higher than HF/6-31G(d,p) is impossible for performance of more accurate calculations. This limitation causes a small difference between both the experimental and theoretical coupling constants in some functional groups.
Table 8: The selected 13C NMR chemical shift (d in ppm) and coupling constants (J in Hz) for some functional groups in the Z–4 and E–4 isomers as a major and minor
| Table 8. The selected 13C NMR chemical shift (d in ppm) and coupling constants (J in Hz) for some functional groups in the Z–4 and E–4 isomers as a major and minor | |||
| groups | dC/ppm | JPC/Hz | |
| 2s 2OMe*
2s 2OMe** |
49.0a (45.2)b , 52.5a (47.0)b *
49.0a (47.0)b , 50.4a (47.0)b** |
||
| d Cipso
d Cipso |
127.0 (126.5)
124.5 (125.6) |
92.3a (92.5)b
91.2a (90.7)b |
|
| d C=O39
d C=O40 |
169.1 (167.3)
169.1 (164.5) |
||
| d Cpara
d Cpara |
133.6 (132.7)
133.1 (133.0) |
||
| d Cmeta
d Cmeta |
128.8 (126.3)
128.5 (126.6) |
||
| d Cortho
d Cortho |
133.6 (133.7)
133.7 (132.7) |
9.2 (10.7)
8.9 (10.1) |
|
| 6C, C6H4
6C, C6H4 |
122.85 (121.3) , 133.5 (137.7)
122.85 (123.2) , 133.4 (127.8) |
||
| NC=O
NC=O |
167.5 (173.3)
167.5 (174.2) |
||
| d, P=C2
d, P=C2 |
38.9 (33.40)
37.2 (31.9) |
||
| d, P-C=C38
d, P-C=C38 |
171.4 (173.3)
170.9 (174.2) |
13.2 (13.0)
13.2 (12.8) |
|
| d, P-C-C36H
d, P-C-C36H |
55.3 (52.7)
54.2 (51.9) |
15.6 (15.53)
15.6 (13.9) |
|
| a Experimental data in accordance with the results reported in the literature [8].
b Theoretical data. * Is relevant to the Z isomer **Is relevant to the E isomer |
|||
Table 9: The selected 1H NMR chemical shift (d in ppm) for some functional groups in the Z–4 and E–4 isomers as a major and minor
| Table 9. The selected 1H NMR chemical shift (d in ppm) for some functional groups in the Z–4 and E–4 isomers as a major and minor | |||
| groups | dH/ppm | ||
| 6H 2s 2CO2Me*
6H 2s 2CO2Me** |
3.78a (3.40)b , 3.80a (3.90)b * 3.71a (2.94)b 3.79a (2.49)b** | ||
| 1H, d, P=C-C-H37
1H, d, P=C-C-H37 |
7.26 (7.07)* 7.26(7.08)
7.33 (7.25) 7.33(7.35)
|
||
| a Experimental data in accordance with the results reported in the literature [8]
b Theoretical data. * Is relevant to the Z isomer **Is relevant to the E isomer |
|||
Conclusions
On the basis of X-ray crystallography data, adjacent carbonyl group in the ylide moiety of synthesized ylide had a resonance with bond of C=P. Also, X-ray study of synthesized ylide exhibited some weak intermolecular C-H…O hydrogen bonds which are effective on stability of the crystal structure. Thus, the results of previous works should be corrected, and the major form should be reported for the Z-isomer of this ylide. Although, dipole moment, number of hydrogen bonds and -Htot in both of the E-4, taken altogether, appeared as two factors on stability of the E-4, but these values are not able to provide a power full effect as a dominance of potential energy. It showed that other factors i.e. other intramolecular exception of H-BCPS and anisotropic effect of phenyl group in ylide 4 containing triphenylphosphine within the structure of the Z-4 isomer could be accounted against mentioned factors for changing the order of relative energy between the Z- and E- isomers. As a result, the Z-4 had a considerable stability with respect to the E-4. As expected, according to theoretical calculations the relative energy at HF/6-31G (d,p) level, were compatible with experimental results from the X-ray crystallography, 1H , 13C and 13P NMR spectroscopy which indicate the Z-4 was more abundance isomer.
Acknowledgments
Authors sincerely thank the University of Sistan and Baluchestan for providing financial support of this work.
References
- Crayson, E. J. Griffith, Topics in Phosphorus Chemistry, Insterscience: New York, vol. 7.1972.
- H. R. Hudson, Primary, Secondary and Tertiary Phosphines, Plyphosphines and Heterocyclic Organophosphorus (ш) Compounds, in the Chemistry of Organophosphorus Compounds, Ed.; Hantley, F. R. Wiley: New York, 1990, Vol. 1, pp. 386-472.
- Z. G. Wang, G. T. Zhang, I. Guzei, J. G.Verkade, J. Org. Chem. 66 (10), 3521 (2001)
- R. Burgada, Y. Leroux, Y. O. El Khoshnieh, Phosphorus Sulfur and Silicon Relat. Elem. 10, 181 (1981)
- M. R. Islami, Z. Hassani, K. Saidi, Synth. Commun. 33, 65 (2003)
- I. Yavari, S. Ali-Asgari, K. Porshamsian, M. Bagheri, J. Sulfur. Chem. 28, 477 (2007)
- I.Yavari, Z. Hossaini, A. Alizadeh, Monatshefte Chemie. 137, 1083 (2006)
- Hazeri, Nourollah; Khorassani, Sayyed M. H.; Maghsoodlou, Malek T.; Marandi, Ghasem; Nassiri, Mahmoud; Shahzadeh, Aqil G, Journal of Chemical Research, Volume 2006, Number 4, 1 April 2006, pp. 215-217(3)
- B. E. Maryanoff, A. B. Reitz, Chem. Rev. 89, 863 (1989)
- A. Ramazani,. A. Bodaghi, Tetrahedron Lett 41, 567 (2000)
- M. Kalantari, M.R. Islami, Z. Hassani, K. Saidi, Arkivoc. (x), 55, 2006.
- Shahraki M, Habibi-Khorassani SM, (2015) J. Phys. Org. Chem. doi: 10.1002/poc.3424
- M. T. Maghsoodlou, N. Hazeri, S.M. Habibi-Khorassani, F. Jalil Milani, M.A. Kazemian, M. Shahraki, Biomedical and Pharamcology Journal, 1(2), 293-296 (2006)
- M. Shahraki, S.M. Habibi-Khorassani, A. Ebrahimi, M. Maghsoodlou, Y. Ghalandarzehi, Struct Chem 2012, 24, 623-635.
- E. Mofarrah, S. M. Habibi-Khorassani, M. T. Maghsoodlou, M. Shahraki, Applied Magnetic Resonance, 2015. 10.1007/s00723-015-0703-2
- S.M. Habibi-Khorassani, M. Shahraki, M.T. Maghsoodlou, S. Erfani, Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy, 2015, 145, 410–416.
- G. M. Sheldrick, Acta Cryst. A64, 112 (2008)
- Oxford Diffraction, CrysAlis CCD. Oxford Diffraction LTD, Abingdon, England. (2006).
- Oxford Diffraction, CrysAlis RED. Oxford Diffraction LTD, Abingdon, England. (2006).
- C. K. Johnson, ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory. Tennessee. (1976).
- L. J. Farrugia, J. Appl. Cryst., 32, 837 (1999).
- H. J. Bestmann, T. Joachim, J. Lengyel, F.Oth. Merenyi, H.Weitkamp, Tetrahedron. Lett. 7, 3225 (1966)
- A. E. Reed, R. B. Eeinstock, F. J. Weinhold, J. Chem. Phys. 83, 735 (1985)
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, J.A. Montgomery, T. Vreven, K.N. Kudin, J.C. Burant, J. M. Millam, S.S. Iyengar, J.Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K.Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R.Gomperts, R. E.Stratmann, O.Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth,Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farka D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople., Gaussian 03. Revision. A.1. Gaussian. Inc.. Pittsburgh PA. (2003)
- R.F. Bader, W. Atoms in molecules A Quantum Theory, Oxford University, New York. 1990
- F. W. Biegler König, J. Schönbohm, D. Bayle, J. Comput. Chem. 22, 545 (2001)
- S. J. Grabowski, J. Mol. Struct. 562, 137 (2001)
- W. D. Arnold, E. Oldfield, J. Am. Chem. Soc. 122, 12835 (2000)
- I. Rozas, I. Alkorta, J. Elguero, J. Am. Chem. Soc. 122, 11154 (2000)
- M. T. H. Liu, Y. N. Romashin, R. Bonneau, Int. J. Chem. Kinet. 26, 1179 (1994)

