
Manuscript accepted on :November 18, 2010
Published online on: 24-11-2015
Plagiarism Check: Yes
G. V. N. Kiranmayi¹, K. Ravi Shankar² and V. Chandrasekhar Nainala²
¹Department of Pharmacy, Sri Vishnu College of Pharmacy, Vishnupur, Bhimawaram - 534202 West Godavari District, A.P., India.
²Sri Sai Aditya Institute of Pharmaceutical Sciences and Research, ADB Road, Surampalem, East Godavari, A.P. India.
³Aditya Institute of Pharmaceutical Sciences and Research, A.P. India.
Abstract
Wilson disease is an inherited, autosomal recessive, copper accumulation and toxicity disorder that affects about 30 individuals per million. This rare disease is caused by mutations in the gene encoding a copper-transporting P-type ATPase, which is important for copper excretion into bile, leading to copper accumulation in the liver. Toxic copper concentrations can also be found in the brain and kidney, and clinical phenotypes include hepatic, haemolytic, neurologic and psychiatric diseases. Diagnosis is based on the combination of clinical features and findings such as increased urinary copper excretion, reduced levels of serum ceruloplasmin, high concentrations of copper in liver tissuesand Kayser–Fleischer rings, biochemical and immunological markers,magnetic resonance imaging ,neuropathological study. Genetic studies are also becoming available for clinical use, but the utility of direct mutation analysis is limited.
Keywords
Wilson disease; new advances; diagnosis; treatment
Download this article as:| Copy the following to cite this article: Kiranmayi G. V. N, Shankar K. R, Nainala V. C. The current Status and New Advances in Diagnosis and Treatment of Wilson Disease. Biomed Pharmacol J 2010;3(2) |
| Copy the following to cite this URL: Kiranmayi G. V. N, Shankar K. R, Nainala V. C. The current Status and New Advances in Diagnosis and Treatment of Wilson Disease. Biomed Pharmacol J 2010;3(2). Available from: http://biomedpharmajournal.org/?p=1554 |
Introduction
Cause
The condition is due to mutations in the Wilson disease protein (ATP7B) gene. A single abnormal copy of the gene is present in 1 in 100 people, who do not develop any symptoms (they are carriers). If a child inherits the gene from both parents, they may develop Wilson’s disease. Symptoms usually appear between the ages of 6 and 20 years, but cases in much older patients have been described. Wilson’s disease occurs in 1 to 4 per 100,000 people.[1] Wilson’s disease is named after Dr Samuel Alexander Kinnier Wilson (1878-1937), the British neurologist who first described the condition in 1912.[2]
Pathophysiology
Copper is needed by the body for a number of functions, predominantly as a cofactor for a number of enzymes such as ceruloplasmin, cytochrome c oxidase, dopamine β-hydroxylase, superoxide dismutase and tyrosinase.[16]
 |
Figure 1:
|
Copper enters the body through the digestive tract. A transporter protein on the cells of the small bowel, copper membrane transporter 1 (CMT1), carries copper inside the cells, where some is bound to metallothionein and part is carried by ATOX1 to an organelle known as the trans-Golgi network. Here, in response to rising concentrations of copper, an enzyme called ATP7A releases copper into the portal vein to the liver. Liver cells also carry the CMT1 protein, and metallothionein and ATOX1 bind it inside the cell, but here it is ATP7B that links copper to ceruloplasmin and releases it into the bloodstream, as well as removing excess copper by secreting it into bile. Both functions of ATP7B are impaired in Wilson’s disease. Copper accumulates in the liver tissue; ceruloplasmin is still secreted, but in a form that lacks copper (termed apoceruloplasmin) and rapidly degraded in the bloodstream.[16]
When the amount of copper in the liver overwhelms the proteins that normally bind it, it causes oxidative damage through a process known as Fenton chemistry; this damage eventually leads to chronic active hepatitis, fibrosis (deposition of connective tissue) and cirrhosis. The liver also releases copper into the bloodstream that is not bound to ceruloplasmin. This free copper precipitates throughout the body but particularly in the kidneys, eyes and brain. In the brain, most copper is deposited in the basal ganglia, particularly in the putamen and globus pallidus (together called the lenticular nucleus); these areas normally participate in the coordination of movement as well as playing a significant role in neurocognitive processes such as the processing of stimuli and mood regulation.
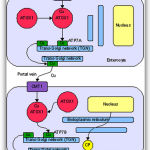 |
Figure 2:
|
It is not clear why Wilson’s disease causes hemolysis, but various lines of evidence suggest that high levels of free (non-ceruloplasmin bound) copper have a direct effect on either oxidation of hemoglobin, inhibition of energy- supplying enzymes in the red blood cell, or direct damage to the cell membrane.[17]
Signs and symptoms
The main sites of copper accumulation are the liver and the brain, and consequently liver disease and neuropsychiatric symptoms are the main features that lead to diagnosis.[1] Patients with liver problems tend to come to medical attention earlier, generally as children or teenagers, than those with neurological and psychiatric symptoms, who tend to be in their twenties or older. Some are identified only because relatives have been diagnosed with Wilson’s disease; many of these patients, when tested, turn out to have been experiencing symptoms of the condition but haven’t received a diagnosis.[18]
Symptoms of excess copper in the liver include
Jaundice ,Swelled abdomen ,Pain in the abdomen ,Nausea ,Vomiting blood Symptoms of excess copper in the brain include:
Depression ,Anxiety , Mood swings .Aggressive or other inappropriate behaviors ,Difficulty speaking and swallowing , Tremors ,Rigid muscles ,Problems with balance and walking Symptoms of excess copper in the eyes:
Kayser-Fleischer rings (rusty or brown-colored ring around the iris)[3,4,5,6]
Liver disease
Liver disease may present as tiredness, increased bleeding tendency or confusion (due to hepatic encephalopathy) and portal hypertension. The latter, a condition in which the pressure on the portal vein is markedly increased, leads to esophageal varices (blood vessels in the esophagus) that may bleed in a life- threatening fashion, splenomegaly (enlargement of the spleen) and ascites (accumulation of fluid in the abdominal cavity). On examination, signs of chronic liver disease such as spider naevi (small distended blood vessels, usually on the chest) may be observed. Chronic active hepatitis has caused cirrhosis of the liver in most patients by the time they develop symptoms. While most people with cirrhosis have an increased risk of hepatocellular carcinoma (liver cancer), this risk is relatively very low in Wilson’s disease.[1]
About 5% of all patients are diagnosed only when they develop fulminant acute liver failure, often in the context of a hemolytic anemia (anemia due to the destruction of red blood cells). This leads to abnormalities in protein production (identified by deranged coagulation) and metabolism by the liver. The deranged protein metabolism leads to the accumulation of waste products such as ammonia in the bloodsteam. When these irritate the brain, the patient develops hepatic encephalopathy (confusion, coma, seizures and finally life-threatening swelling of the brain).[1]
 |
Figure 3:
|
Neurological features
Neurological disorders usually develop in the third decade of life and are the presenting symptoms of Wilson disease in 40–50% of patients [11]. Typically, these involve hypokinetic speech, tremor, dystonia, incoordination and dysphagia; the patient may appear to have Parkinson’s disease because the copper typically accumulates in the basal ganglia. Kayser Fleischer rings are observable in virtually all patients with neurological involvement [12].
In 2005, a score was developed to describe the neurological signs and symptoms commonly found in Wilson disease patients [13]. The most representative and easiest to apply for the purpose of a retrospective neurological assessment were: six signs (rigidity, bradykinesia, ataxia, tremors, dyskinesia, dystonia) and four functions (eating, walking, talking, self care and dressing). Each item scores a maximum of 3, higher scores coinciding with milder symptoms or less-impaired functions. The maximum score is 30. The score is mainly of descriptive value and particularly useful for retrospective analysis; it can be used not only by neurologists, but also by gastroenterologists or hepatologists.
Psychiatric features
Psychiatric symptoms can occur before (most likely 2 or 3 years before) hepatic and neurological symptoms become manifest. Patients may develop psychiatric and behavioural abnormalities, e.g. depression (sometimes leading to attempted suicide), paranoia, hallucinations and delusions, irritability, loss of sexual inhibitions or declining performance at school or at work [14]. Behavioural and cognitive symptoms can be reversed by 1–2 years of continuous treatment [15].
Other organ systems
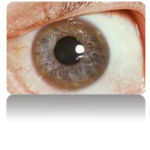 |
Figure 4:
|
A Kayser-Fleischer ring in a patient with symptoms suggestive of Wilson’s disease
Various medical conditions have been linked with copper accumulation in Wilson’s disease:
Eyes: Kayser-Fleischer rings (KF rings) may be visible around the iris. They are due to copper deposition in Descemet’s membrane of the cornea. They do not occur in all patients and may only be visible on slit lamp examination. Wilson’s disease is also associated with sunflower cataracts, brown or green pigmentation of the anterior and posterior lens capsule. Neither cause significant visual loss.[1] KF rings occur in 66% of cases, more often in those with neurological than with liver problems.[18]
Kidneys: renal tubular acidosis, a disorder of bicarbonate handling by the proximal tubules leads to nephrocalcinosis (calcium accumulation in the kidneys), weakening of the bone (due to calcium and phosphate loss) and occasionally aminoaciduria (loss of amino acids, needed for protein synthesis).[1]
Heart: cardiomyopathy (weakness of the heart muscle) is a rare but recognized problem in Wilson’s disease; it may lead to heart failure (fluid accumulation due to decreased pump function) and cardiac arrhythmias (episodes of irregular and/or abnormally fast or slow heart beat).[1]
Hormones: hypoparathyroidism (failure of the parathyroid glands, leading to low calcium levels), infertility and habitual abortion.[1]
Diagnosis
The laboratory diagnosis of WILSON DISEASE can be broadly classified as : biochemical , ophthalmological, electrophysiological and radiological .each of these investigations may have varied diagnostic and prognostic significance.
Biochemical
Under this the following tests like estimation of 24-h urinary copper excretion , Hepatic copper concentration , Serum copper concentration , Ceruloplasmin concentration are done .
24-h urinary copper excretion
A urinary copper concentration greater than 100 _g/24 h (>1.6_mol/24 h) is considered diagnostic for Wilson disease. This level is reached in most symptomatic patients, but a level of 40–100 _g does not enable Wilson disease to be ruled out in asymptomatic patients, so they require further testing [23]. In individuals who are heterozygous for Wilson disease, moreover, urinary copper excretion is rarely above 70_g/24 h. The utility of this measurement is further limited by the fact that high urinary copper levels can be found in other chronic liver diseases too (e.g. primary biliary cirrhosis, primary sclerosing cholangitis, Alegille syndrome and autoimmune hepatitis) [24].
Estimating urinary copper excretion may also be misleading due to improper of the 24-h urine collection or to copper contamination. Preservatives used for routine analysis may contain mercuric oxide i.e. Stabilur), which interferes with all metal testing. If both urinalysis and metal testing are ordered, a separate urine specimen9containing no additive) has to be used for the metal testing. Testing urinary copper excretion after penicillamine challenge can be useful, though this test has only been standardized
in a pediatric population [25], and it has never been validated in heterozygous carriers of Wilson disease. This test involves administering 500 mg of penicillamine at the baseline and then again 12 h after starting the 24 h urine collection. If the final urinary copper concentration is greater than 1600_g/24 h, then a diagnosis ofWilson disease is more likely than other liver diseases (e.g. autoimmune chronic active hepatitis, primary sclerosing cholangitis or acute liver failure) [25]
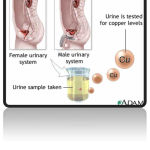 |
Figure 5:
|
The copper urine test is used to determine the presence of Wilson disease, a sometimes fatal condition in which the buildup of excess copper damages the liver, and eventually the kidneys, eyes and brain.
Hepatic copper concentration
In almost all patients, the liver’s copper content is more than 250_g/g dry weight (normal <50 _g/g dry weight) and may even be as high as 3000 _g/g dry weight [26]. Patients with severe cirrhosis occasionally have a liver copper content below 250 _g/g dry weight because of an uneven distribution of copper in the liver parenchyma. The hepatic copper concentration is reportedly lower than 250_g/g dry weight in up to 20% of Wilson patients, especially among those with mainly neuropsychiatric involvement [27,28]. These results derive from our limited experience on 35 patients [28] and from Ferenci’s study on 114 patients [27], whose hepatic copper was measured at different centers and in different conditions.In addition, the variability of hepatic copper may also be due to sampling error or the poor reliability of a single copper determination. Measuring hepatic copper nonetheless remains the best diagnostic test. Histochemical methods for detecting excess copper in the liver are unreliable[29].
Serum copper concentration
The plasma free copper concentration (i.e. copper not bound to ceruloplasmin) is considered useful for the diagnosis of Wilson disease, but it can only be calculated after measuring the total concentration of copper and ceruloplasmin in plasma. Since the amount of copper bound to ceruloplasmin is about 3.15 _g/mg of ceruloplasmin, the serum free copper concentration is calculated as the difference between the serum copper concentration (_g/dL) and three times the ceruloplasmin concentration (mg/dL) [30]. The serum free copper concentration is more than 25 _g/dL in most untreated patients (normal value <15 _g/dL), but it may also be higher than normal in patients with acute liver failure or chronic cholestasis [31] and its validity as a test depends on how well ceruloplasmin was measured (the enzymatic method appears to be advisable, though not universally accepted) [32]. This test is probably more useful during the follow-up, to assess response to treatment, than as a single diagnostic tool.
Ceruloplasmin concentration
 |
Figure 6:
|
Ceruloplasmin is a copper-carrying protein that is bound to 90% of the circulating copper in normal individuals. The normal concentration of ceruloplasmin is 200–400 mg/L, and a serum ceruloplasmin level below 200 mg/L (20 mg/dL) is suggestive of Wilson disease. The clinical utility of this measurement is limited, however, by the fact that ceruloplasmin concentrations under 200 mg/L can be found in 1% of controls, in 10% of heterozygous Wilson disease carriers and in patients with copper deficiency [33], Menkes disease, hereditary hypoceruloplasminemia [34], malabsorption, nephrotic syndrome and chronic liver failure. What’s more, normal ceruloplasmin concentrations are recorded in about 20% of Wilson disease patients. There is still some debate as to whether the immunological-nephelometric method is comparable with enzymatic assay (oxidase activity) for determining ceruloplasmin serum concentration, especially in patients with liver disease. Enzymatic activity is the biologically relevant parameter, whereas the immunonephelometric method measures both ceruloplasmin and the biologically inactive apo-form. The enzymatic method should consequently be preferred for the diagnosis of Wilson disease [35], though a weak correlation was found between ceruloplasmin protein concentration and oxidase activity in patients with different degrees of hepatic involvement [32]. A more recent study on 33Wilson patients demonstrated a good correlation between the two tests [36].
Ohpthalmological
The presence of Kayser–Fleischer rings indicates that free copper has been released into the circulation. Other ophthalmological findings include sunflower cataracts, which are the sign of copper deposition in the lens. Kayser–Fleischer rings are seen in 50–60% of patients with mainly hepatic disease, whereas almost all patients with mainly neuropsychiatric symptoms are positive for ophthalmological signs [37]. Kayser–Fleischer rings are not found in all Wilson disease patients, however, and they are not completely specific for the disorder—they can occur in patients with chronic cholestatic diseases and in neonatal cholestasis [38]. The rings may disappear after long-term therapy, though their presence does not correlate with disease severity[39].
Genetic testing
Given the variability of the biochemical and clinical features of Wilson disease, mutation analysis is becoming more and more essential to confirm a suspicion of the disorder. Nearly 300 ATP7B mutations have been identified to date. When mutations responsible forWilson disease are detected, the condition can be confirmed, but a negative result cannot exclude a diagnosis ofWilson disease. Some populations had proved to have a limited number of predominant mutations, such as in Eastern Europe (H1069Q) [40], Sardinia (c-441 427del15) [41], Korea (Arg778Leu) [42], Iceland (2007del7) [43], Japan (229insC, Arg778Leu) [44], Spain (Met645Arg) [45] and China (Arg778Leu) [46]. In these geographical areas, utilization of mutation analysis may be a useful diagnostic method. For the time being, genetic testing is mainly limited to the screening of first-degree relatives of Wilson disease patients, since haplotype analysis is excellent for genotyping the full siblings of an index case [47].
While it is true that a negative result cannot rule out a diagnosis of Wilson disease, many laboratories now sequence the complete gene as well as the promoter. The availability of this technique is limited, and standardization is not well-established.
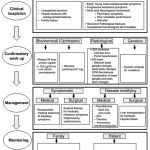 |
Figure 7:
|
Wilson’s disease has an autosomal recessive pattern of inheritance.
The Wilson’s disease gene (ATP7B) has been mapped to chromosome 13 (13q14.3) and is expressed primarily in the liver, kidney, and placenta. The gene codes for a P-type (cation transport enzyme) ATPase that transports copper into bile and incorporates it into ceruloplasmin.[1] Mutations can be detected in 90% of patients. Most (60%) are homozygous for ATP7B mutations (two abnormal copies), and 30% have only one abnormal copy. 10% have no detectable mutation.[18]
Although 300 mutations of ATP7B have been described, in most populations the cases of Wilson’s disease are due to a small number of mutations specific for that population. For instance, in Western populations the H1069Q mutation (replacement of a histidine by a glutamine at position 1069 in the gene) is present in 37-63% of cases, while in China this mutation is very uncommon and R778L (arginine to leucine at 778) is found more often.
Relatively little is known about the relative impact of various mutations, although the H1069Q mutation seems to predict later onset and predominantly neurological problems, according to some studies.[1][16]
A normal variation in the PRNP gene can modify the course of the disease by delaying the age of onset and affecting the type of symptoms that develop. This gene produces prion protein, which is active in the brain and other tissues and also appears to be involved in transporting copper.[21] A role for the ApoE gene was initially suspected but could not be confirmed.[16]
The condition is inherited in an autosomal recessive pattern, which means both copies of the gene have mutations. In order to inherit it, both of the parents of an individual must carry an affected gene. Most patients have no family history of the condition.[16] People with only one abnormal gene are called carriers (heterozygotes) and may have mild, but medically insignificant, abnormalities of copper metabolism.[20]
Wilson’s disease is the most common of a group of hereditary diseases that cause copper overload in the liver. All can cause cirrhosis at a young age. The other members of the group are Indian childhood cirrhosis (ICC), endemic Tyrolean infantile cirrhosis and idiopathic copper toxicosis. These are not related to ATP7B mutations, but ICC has been linked to mutations in the KRT8 and the KRT18 gene.[16]
Electro physiological
Electrophysiological studies have been done in Wilson Disease to document any subclinical abnormalities and as such do
Neurophysiological evaluation
Only a few studies have reported electrophysiological studies such as electroencephalography (EEG), visual- evoked potentials, brain stem auditory evoked potentials, somatosensory-evoked potentials and nerve conduction studies. [ 19 ,4 8 , 49 ] In our study, the reported abnormalities included: EEG-41.1%, visual-evoked potentials- 35% and brain stem auditory evoked potentials-42.1%[ 19 ]. Earlier Satishchandra etal had found subclinical involvement of optic nerve and caudal brain stem auditory pathways. [ 4 8 ]
In another study electrophysiological evaluation to study the visual pathway abnormalities in WILSON DISEASE was carried out using electroretinography and visual-evoked potentials and has demonstrated reversibility of retinal dysfunction with clinical improvement. [ 5 0 ]
Autonomic nervous system involvement has also been From the reported in WILSON DISEASE . In a study, six of the 13 patients with electrophysiological evidence of dysautonomia had also clinical evidence of dysautonomia ,it was concluded that subclinical dysautonomia in Wilson Disease perhaps
that subcli nical dysautonomi a in WILSON DISEASE is perhaps due to central autonomic dysregulation involving both sympathetic and parasympathetic systems.[51 ] Abnormalities of cardiovascular reflexes have also been reported[ 5 2 ]. While majority of these patients are asymptomatic baring few exceptions. [ 5 3 , 5 4 ] In Indian literature, the only publication has included various electrocardiographic changes in WILSON DISEASE . Fifteen among the 50 patients were noted to have an abnormality and the ECG included sinus tachycardia, sinus bradycardia , bifid d P wave, ST elevation, ST depression, T wave inversion , ventricular premature e contraction on and prominent t U waves.[55] The authors concluded that these abnormalities were probably related to copper deposition in the myocardium.
Imaging
Imaging studies in Wilson Disease have been carried over years an d included X-rays, CT brain , MR imaging and spectroscopy.[56-63]
Roentgenograms
The various skeletal abnormalities in our study included osteoporosis (38.8%); fractures (3.4%); arthritis (4.1% and milkman pseudo fractures (8.2%). In our study, renal stones or gallstones were detected by ultrasonography and/or X-rays in 4.9 and 2.8% patients, respectively.
Computed tomography (CT brain)
Prior to widespread ad availability of MRI facilities CT scan was the imaging modality of the brain However CT grossly underestimates the pathology of Wilson Disease . In a series of 116 patients of WILSON DISEASE , the observed CT abnormalities included: Cortical atrophy (44.8%) ventricular dilatation (44%), caudate atrophy (25%),brain stem atrophy (31.9%), cerebellar atrophy (19%) and hemispheric (29.3%), basal ganglionic (19.8%), thalamic(10.3%) hypo densities [1,56]
Magnetic resonance imaging of brain
Introduction of MR imaging has greatly helped the clinicians to understand the pathological and anatomical correlates of clinical manifestations in Wilson Disease . MRI of the brain can assist in the diagnosis and may also help in prognostication. In a large study of MRI in 100 patients with WILSON DISEASE , the salient findings included: Atrophy of the cerebrum (70%), brainstem (66%) and cerebellum(52%), signal abnormalities in put amen (72%), caudate(61%), thalami (58%), m midbrain (49%), Pons (20%), cerebral white matter (25%), cortex (9%), medulla (12%) and cerebellum (10%). The characteristic ‘face of giant panda’ sign was noted in 12% and feature of central pontine myelinolysis was noted in 7% and bright claustral sign i n 4% of patients. [5 7 ] from the same center, sequential MRI changes during the course of the treatment were studied in 50 patients. In this study the imaging features had improved in 70% of patients, not changed in 20%, worsened in 8% and the changes were both improvement and worsening in 2% of patients, improvement in imaging features, 20%; had no significant changes, 8%; had worsening of imaging features in 2%. These observations suggest that MRI can be used as a tool to monitor the therapy in WILSON DISEASE [58]The important MRI features that distinguish WILSON DISEASE from other movement disorders are the presence of central pontine myelinolysis-like changes and midbrain tectal plate signal changes[ 59 , 6 4 ]
Magnetic resonance spectroscopy provides insight into the biochemical changes in Wilson Disease . Study of MRS of basal ganglia showed reduced breakdown and/or increased synthesis of membrane phospholipids and increased neuronal damage in patients with WILSON DISEASE [ 6 5 ]
Therapy
The goal of therapy is to reduce copper accumulation, either by enhancing its urinary excretion or by reducing its intestinal absorption. The options available for the treatment of Wilson disease are all the more effective the sooner they are initiated.
D-Penicillamine
Penicillamine (d-PCA) mobilizes copper and forms copper–penicillamine complexes that are excreted in the uri Most patients with a mainly hepatic Wilson disease phenotype experience a clinical improvement after 6–8 weeks of treatment, but it may take 6–12 months for the change to be noticeable [66]. It is best to start patients on lower doses, gradually increasing it up to the therapeutic range in order to improve its tolerability [69]. For instance, d-PCA treatment is started at 125 mg/day for the first week, then the dose is raised by 125 mg every week up to a dose of 1.0–1.5 g/day. d-PCA can induce vitamin B6 deficiency so a daily dose of 25 mg of pyridoxine (vitamin B6) is usually added to the treatment regimen. Approximately 30% of patients have hypersensitive reactions in the first month of treatment (fever, rash, lymphadenopathy) [67]. These early side effects are usually transient, but temporary drug withdrawal and corticosteroids may be required. Late drug reactions can be seen even after years of uneventful treatment. The most common of these late side effects involve the skin (degenerative changes, elastosis perforans serpiginosa) [68] and joints (arthropathy), or may be mediated by immunological effects (lupus-like reactions, nephrotic syndrome, myas thenia gravis, Goodpasture syndrome) [67]. Bone marrow depression, which can present as aplastic anaemia, neutropenia and thrombocytopenia, may develop as an early or late side effect, so a full blood count is needed before the treatment is started. Neurological symptoms reportedly become more severe in about 50% of patients taking d- PCA [70].
Trientine
Trientine is a copper chelator, taking effect primarily by enhancing urinary copper excretion. It is used as an alternative to d-PCA, both in the event of tolerance and as front-line treatment because it has fewer side effects. It is thought to act by mobilizing tissue copper, albeit to a lesser degree than d-PCA (hence the more limited side effects) [71]. The usual starting dose of trientine is 750–1500 mg, divided between 2–3 doses a day, with 750– 1000 mg as a maintenance dose. It should be taken before meals or 2 h afterwards. Trientine is probably less toxic than d-PCA. No early hypersensitivity reactions have been reported; late adverse reactions include lupus- like syndrome with proteinuria and moderate sideroblastic anaemia [72]. The risk of neurological symptoms becoming worse when trientine is used as first-line therapy is reportedly 26%[73].
Zinc
Zinc increases the levels of intestinal cell metallothionein [74,75], a protein with a strong affinity for copper. The tight link formed between copper and metallothionein inhibits further copper absorption and promotes its loss in the faeces as enterocytes are shed due to normal cell turnover. Zinc may also increase metallothionein levels in hepatocytes, i.e. the copper binding to metallothionein forms non-toxic complexes in the liver, resulting in little or no change in the total hepatic copper concentration [76]. Zinc is used mainly as a front-line therapy in patients who have not (or not yet) developed symptoms [77], for maintenance therapy and for patients with a mainly neuropsychiatric involvement, because aworsening of the neurological picture is very uncommon in such cases (10% in our experience) [86]. Zinc sulfate should be administered at a dose of 220 mg/day three times daily (corresponding to 150 mg of elemental zinc a day), at least 1 h before meals [78]. Zinc is generally well tolerated. Gastric irritation is the most frequent problem (10–15%) [78], but this can be overcome by replacing zinc sulfate with zinc acetate, or by taking the first daily dose mid-morning rather than before breakfast. A mild, harmless increase in serum amylase and lipase concentrations [79] (that is not due to pancreatitis), a 20% reduction of high-density lipoprotein cholesterol in male patients [80] (compensated by a reduction in total cholesterol) have been described.
Combination therapies
The combined use of d-PCA and zinc is not recommended,as it does not make sense to administer a metal with a chelating agent capable of neutralizing its effect. A previous study [81] failed to demonstrate any advantage of such a combined treatment. A highly experimental approach involves using trientine combined with zinc for 6–8 weeks, then switching to zinc maintenance therapy. This strategy was used to treat nine patients presenting with hepatic decompensation [82], whose liver function improved, with a normalization of their Child–Turcotte–Pugh score, and even a markedly reduced liver fibrosis. If doses of chelators and zinc are to be administered in combination, the interval between them must be as wide as possible; this means taking medication numerous times during the day and this is very likely to have a negative effect on compliance.
Tetrathiomolybdate
Tetrathiomolybdate has two mechanisms of action [83].First, it complexes copper in the intestinal lumen, preventing its absorption. Second, once it has been absorbed, it complexes copper with albumin in the blood and makes the copper unavailable for cellular uptake. Tetrathiomolybdate has been proposed as initial treatment for patients with neurological signs and symptoms [84], but its use is restricted by the limited clinical experience with the drug and it is not commercially available in the USA or the European Union. Neurological Wilson disease patients have been treated with doses of tetrathiomolybdate varying from 120 to 410 mg/day for 8 weeks [84]. A randomized, double-blind, controlled, two-arm study on 48 Wilson disease patients with a neurological presentation was recently performed: patients were treated with trientine (500 mg twice a day) or tetrathiomolybdate (20 mg three times a day with meals and 20 mg three times a day between meals) for 8 weeks [73]. Fewer patients on tetrathiomolybdate experienced a neurological deterioration than those on trientine, and about 15% of patients on tetrathiomolybdate experienced only mild side effects, which included bone marrow toxicity (anaemia, thrombocytopenia and neutropenia) and rising aminotransferase levels, but both effects were reportedly transient and responded to suspension of the drug.We reported on a case of acute hepatitis with high transaminases, signs of cholestasis and a marked increase in cholesterol and triglycerides [85] after tetrathiomolybdate treatment in a neurological Wilson patient, whose neurological functions remained stable throughout the course of therapy.
Diet
Foods rich in copper (e.g. liver, chocolate, nuts, mushrooms and shellfish) should be avoided, at least in the early years after diagnosis. More stringent dietary measures are unpleasant, impractical and probably fail to postpone the progression of disease [87]. Drinking water usually contains less than 0.2 mg copper per liter but up to 10% of domestic drinking water has copper levels that may be too high for Wilson patients, so it should be tested.
Liver transplantation
Liver transplantation is the ultimate treatment for patients with Wilson disease. Survival rates reportedly range from 100% at 33 months [88] to 62% at 1 year [89]. We have reported overall patient survival rates at 6 and 12 months and 5 and 10 years after transplantation of 89.1, 89.1, 75.6, 58.8%, respectively [95]. Wilson disease patients should be considered for liver transplantation when suitable medical therapy has failed or in the case of acute liver failure, when there is no time for other therapies to take effect. Patients with a combination of hepaticand neuropsychiatric conditions warrant careful neurological assessment, but liver transplantation is contraindicated only in cases of severe neurological impairment. Neuropsychiatric symptoms are always a contraindication for liver transplantation [90-92]. A prognostic scoring system for Wilson disease was recently developed [93], after the revision of a previous scoring system proposed in 1986 [94]. The new scoring system was developed for pediatric patients and is based on bilirubin, AST and albumin levels, white cell count and INR. A score of 11 or above suggests that patients at high risk of mortality unless liver transplantation is performed. This score revealed a sensitivity of 93% and a specificity of 97%, with a positive predictive value of 92%.
Wilson disease in pregnancy
Pregnancy is not contraindicated for patients with well managed Wilson disease and compensated liver disease, but must be continued throughout pregnancy and breast feeding because its interruption could lead to acute liver failure. safest drugs for pregnant Wilson disease patients are zinc and trientine [96]. A report from Brewer et al. [97] described 26 pregnancies in 19 women on zinc therapy throughout their pregnancy, showing that zinc protected the health of the mother and foetus during the pregnancy, with a 7.7% rate of congenital defects (one baby was born with a heart defect, another had microcephaly and died an hour after birth), as opposed to the 4% rate of major and minor congenital defects in the general population, but studies on larger groups would be needed to ascertain whether the rate of defects in zinc-treated pregnancies is actually higher than in the general population. It has also been demonstrated that zinc does not reach significantly high concentrations in breast milk [98]The teratogenicity of d-PCA is still debated: while some authors believe there is no reason for concern regarding use of d-PCA in pregnancy [99]cutis laxa syndrome has been reported in about 5% of babies born from mothers treated with d-PCA during pregnancy [100], as well as other severe embryopathies (micrognathia, contractures of all limbs, central nervous system abnormalities) [101].
Medici et al. / Digestive and Liver Disease 39 (2007) 601–609 607
Conclusions
Wilson disease is a rare inherited metabolic disease leading to copper accumulation, mainly in the liver and brain. Although this accumulation of copper begins at birth, symptoms of the disorder usually appear later in life, between the ages of 3 and 40. The primary consequence for approximately 40% of Wilson patients is liver disease. In other patients, the first symptoms are neurological or psychiatric, or both. Without proper treatment, Wilson disease is generally fatal, usually by the age of 30. If treatment is begun early enough, symptomatic recovery is usually complete, and a normal length and quality of life can be expected. Even decompensated liver disease usually improves with adequate therapy.
In most cases, a well-timed treatment can overcome both the hepatic and the neurological signs. The main goal of research should consequently be early diagnosis for both hepatic and neurological Wilson disease, as well as new accurate methods for ensuring compliance with the treatment.
References
- Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML (2007). “Wilson’s disease”. Lancet 369 (9559): 397–40 doi:10.1016/S0140-6736(07)60196-2. PMID 17276780.
- Kinnier Wilson SA (1912). “Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver” (PDF). Brain 34 (1): 295–507. doi:10.1093/brain/34.4.295
- https://wwaasld.org/
- http://wwwilsonsdisease.org/
- http://wwliver.ca/
- http://wwhc-sc.gc.ca/
- Yarze JC, Martin P, Munoz SJ, Friedman LWilson’s disease: current status. Am J Med 1992;92:643– 54.
- Brewer GJ, Yuzbasiyan-Gurkan V. Wilson dise Medicine 1992; 71:139–64.
- Medici V, Mirante VG, Fassati LR, Pompili M, Forti D, Del Gaudio M, et al. Liver transplantationfor Wilson’s disease: the burden of neuropsychiatric disord Liver Transpl 2005;11:1056–63.
- Brewer GJ. Recognition, diagnosis, and management of Wilson’s disease. PSEBM 2000;223:39–
- Akil M, Brewer GJ. Psychiatric and behavioral abnormalities in Wilson’s disease Adv Neurol 1995;65:171–
- de Bie P, Muller P, Wijmenga C, Klomp LW (November 2007). “Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes“. J. Genet. 44 (11): 673–88. doi:10.1136/jmg.2007.052746. PMID 17717039.
- Lee, GR (1999). “Chapter 48: acquired hemolytic anaemias resulting from direct effects of infectious, chemical or physical agents”, in Lee GR, Foerster J, Lukens J et al: Wintrobe’s clinical hematology, 10th, Williams & Wilkins, 12 ISBN 0-683-18242-0.
- Merle U, Schaefer M, Ferenci P, Stremmel W (2007). “Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study“. Gut 56 (1): 115–20. doi:10.1136/gu2005.087262. PMID16709660.
- Taly AB, Meenakshi-Sundaram S, Sinha S, Swamy HS, Arunodaya Wilson disease: Description of 282 patients evaluated over 3 decades. Medicine (Baltimore) 2007;86:112-21.
- Roberts EA, Schilsky ML (2003). “A practice guideline on Wilson disease” (PDF). Hepatology 37 (6): 1475– doi:10.1053/jhep.2003.50252. PMID 12774027.
- ↑ Grubenbecher S, Stüve O, Hefter H, Korth C (2006). “Prion protein gene codon 129 modulates clinical course of neurological Wilson disease”. Neuroreport 17 (5): 549–52. doi:11097/01.wnr.0000209006.48105.90. PMID 16543824.
- Steindl P, Ferenci P, Dienes HP, Grimm G, Pabinger I, Madl C, et Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 1997;113:212–8.
- Frommer Urinary copper excretion and hepatic copper concentrations in liver disease. Digestion 1981;21:169–78.
- Martins da Costa C, Baldwin D, Portmann B, Lolin Y, Mowat AP, Mieli-Vergani Value of Urinary copper excretion after penicillamine challenge in the diagnosis of Wilson’s disease. Hepatology 1992;15:609–15.
- Hoogenraad Diagnosis. in: Intermed Medical Publishers. Wilson’s disease. 2nd ed: Amsterdam- Rotterdam; 2001.p. 109–137.
- Ferenci P, Steindl-Munda P, Vogel W, Jessner W, Gschwantler M, Stauber Diagnostic value of quantitative copper determination in patients with Wilson’s disease. Clin Gastroenterol Hepatol 2005;3: 811–8.
- Medici V, Trevisan CP, D’Inc`a R, Barollo M, Zancan L, Fagiuoli S, et a Diagnosis and management of Wilson’s disease: results of a single center experience. J Clin Gastroenterol 2006;40:936–41.
- Pilloni L, Lecca S, Van Eyken P, Flore C, Demelia L, Pilleri G, et Value of histochemical stains for copper in the diagnosis of Wilson’s disease. Histopathology 1998;33:28–33.
- Gaffney D, Fell GS, O’Reilly GaffneyWilson’s disease: acute and presymptomatic laboratory diagnosis and monitoring. J Clin Pathol 2000;53:807–12.
- Gross Jr JB, Ludwig J, Wiesner RH, McCall JT, LaRusso Abnormalities in tests of copper metabolism in primary sclerosing cholangitis. Gastroenterology 1985;89:272–8.
- Macintyre G, Gutfreund KS, Martin WR, Camicioli R, Cox DW. Value of an enzymatic assay for the determination of serum ceruloplasmi J Lab Clin Med 2004;144:294–301.
- Fuhrman MP, Herrmann V, Masidonski P, Eby Pancytopenia after removal of copper from total parenteral nutrition. J Parenteral Enteral Nutr 2001;24:361–6.
- Edwards CQ, Williams DM, Cartwright Hereditary hypoceruloplasminemia. Clin Genet 1979;15:311–6.
- Walshe Wilson’s disease: the importance of measuring serum caeruloplasmin non immunologically. Ann Clin Biochem 2003;40: 155–221.
- Gnanou JV, ThykadavilVG, ThuppilV. Pros and cons of immunochemical and enzymatic method in the diagnosis ofWilson’s diseas Indian J Med Sci 2006;60:371–5.
- Gow PJ, Smallwood RA, Angus PW, Smith AL, Wall AJ, Sewell RB. Diagnosis of Wilson’s disease: an experience over three deca Gut 2000;46:415–9.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman Kayser– Fleischer rings in patients without Wilson’s disease. Gastroenterology 1977;72:1331–5.
- Esmaeli B, Burnstine MA, Martonyi CL, Sugar A, Johnson V, Brewer GJ. Regression of Kayser– Fleischer rings during oral zinc therapy: correlation with systematic manifestations ofWilson’s disease. Cornea 1996;15:582–8.
- Caca K, Ferenci P, Kuhn HJ, Polli C, Willgerodt H, Kunath B, et a High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol 2001;35:575–81.
- Loudianos G, Dessi, Lovicu M, Angius A, Figus A, Lilliu F, et Molecular characterization of Wilson disease in the Sardinian population-evidence of a founder effect. Hum Mutat 1999;14:294–303.
- Kim EK, Yoo OJ, Song KY, Yoo HW, Choi SY, Cho SW, et Identification of three novel mutations and a high frequency of the Arg778Leu mutation in Korean patients with Wilson disease. Hum Mutat 1998;11:275–8.
- Thomas GR, Jensson O, Gudmundsson G, Thorsteinsson L, Cox DW. Wilson disease in Iceland: a clinical and genetic study.AmJHumGenet 1995;56:1140–6
- Nanji MS, Nguyen VT, Kawasoe JH, Inui K, Endo F, Nakajim T, et Haplotype and mutation analysis in Japanese patients with Wilson disease. Am J Hum Genet 1997;60:1423–9.
- Margarit E, Bach V, Gomez D, Bruguera M, Jara P, Queralt R, et Mutation analysis of Wilson disease in the Spanish population— identification of a prevalent substitution and eight novel mutations in the ATP7B gene. Clin Genet 2005;68:61–8.
- Wu ZY, Wang N, Lin MT, Fang L, Murong SX, Yu Mutation analysis and the correlation between genotype and phenotype of Arg778Leu mutation in Chinese patients with Wilson disease. Arch Neurol 2001;58:971–6.
- Roberts EA, Schilsky A practice guideline on Wilson disease. Hepatology 2003;37:1475–92.
- Satishchandra P, Swamy H Visual and brain stem auditory evoked responses in Wilson’s disease. Acta Neurol Scand 1989;79:108-13.
- Das M, Misra UK, Kalita J. A study of clinical, MRI and multimodality evoked potentials in neurolog ic Wilson disease. Eur J Neurol 2007;14:498-50
- Satishchandra P, Ravishankar Naik K. Visual pathway abnormalities Wilson’s disease: An electrophysiological study using electroretinography and visual evoked potentials. J Neurol Sci 2000;176:13-20
- Meenakshi-Sundaram S, Taly AB, Kamath V, Arunodaya GR, Rao S, Swamy HS. Autonomic dysfunction in Wilson’s disease —a clinical and electrophysiological study. Clin Auton Res 2002;12:185-
- Bhattacharya K, Velickovic M, Schilsky M, Kaufmann Autonomic cardiovascular reflexes in Wilson’s disease. Clin Auton Res 2002;12:190-2.
- Kumar Severe autonomic dysfunction as a presenting feature of Wilson’s disease. J Postgrad Med 2005;51:75-6.
- Kuan P. Cardiac Wilson’s disease. Chest 1987;91:579-8
- Meenakshi-S undaram S, Sinha S, Rao M, Pra sha nth LK Arunodaya GR, Rao S, et a l Cardiac involvement in Wilson’s disease—An electrocardiographic observation.J Assoc Physicians India 2004;52:294-
- Bhattacharya D . Follow up study of CT scan b rain and clinical correlation in Wilson’s disease. Paper submitted to NIMHANS in partial fulfillment of requirement for the award of DM degree in neurology Deemed University, NIMHANS-1996.
- Sinha S, Taly AB, Ravishankar S, Prashanth LK, Venugopal KS Arunodaya Wilson’s disease: Cranial MRI observations and clinical correlation. Neuroradiology 2006;48:613-21.
- Sinha S, Taly AB, Prashanth LK, Ravishankar S, Arunodaya GR Vasudev MK. Sequential MRI changes in Wilson’s disease with de coppering therapy: A study of 50 patients. Br J Radiol 2007;80:744-
- Sinha S, Taly AB, Ravishankar S, Prashanth LK, Vasudev MK. Central pontine signal changes in Wilson’s disease: Distinct MRI morphology and sequential changes with de- coppering therapy. J Neuroimaging 2007;17:286-9
- Saha P, Jain S, Mishra NK, Khosla A, Maheshwari M Extensive CT scan abnormality in Wilson’s disease. J Assoc Physicia ns India 1991;39:568-9.
- Sankhyan N, Sharma S, Kalra V, Garg A, Balkrishnan P. Cystic white-matter changes in childhood Wilson’s diseas Pediatr Neurol 2008;39:281-2
- Thapa R, Ghosh A. ‘Face of the giant panda’ sign in Wilson’s disease Pediatr Radiol 2008;38:1355
- Shivakumar R, Thomas S Teaching NeuroImages: Face of the giant panda and her cub: MRI correlates of Wilson disease. Neurology 2009;72: E50
- Prashanth LK, Sinha S, Taly AB, Vasudev MK. Do MRI Features distinguish Wilson’s disease from other early onset extrapyramidal disorders? An analysis of 100 cases. Movement Disorders 2009 (in press).
- Sinha S, Ravishankar S, Taly AB, Arunodaya GR, Prashanth LK, Vasudev M Wilson’s disease: AP and H MR spectroscopic study Neurology 2004;62:539-40.
- Czlonkowska A, Gajda J, Rodo M. Effects of long term treatment in Wilson’s disease with D- penicillamine and zinc sulphate. J Neurol 1996;243:269–73.
- Scheinberg IH, Sternlieb I. Wilson’s disease. In: Smith Jr LH, Major problems in internal medicine. Philadelphia:W.B. Saunders Company; 1984. p. 23–34.
- Hill VA, Seymour CA, Mortimer PS. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa inWilson’s disease. Br J Dermatol 2000;142:560–
- Hoogenraad Diagnosis. in: Intermed Medical Publishers. Wilson’s disease. 2nd ed:Amsterdam – Rotterdam2001.p. 109–137.
- Brewer GJ, Terry CA, Aisen AM, Hill Worsening of neurologic syndrome in patientswith Wilson’s disease with initial penicillamine therapy. Arch Neurol 1987;44:490–3.
- Scheinberg IH, Jaffe ME, Sternlieb The use of trientine in preventing the effects of interrupting penicillamine therapy inWilson’s disease. N Engl J Med 1987;317:209–13.
- Walshe JM. Treatment of Wilson’s disease with trientine (triethylene tetramine) dichlori Lancet 1982;1:643–7.
- Brewer GJ, Askari F, Lorincz MT, Carlson M, SchilskyM, Kluin KJ, et Treatment of Wilson disease with ammonium tetrathiomolybdate. IV. Comparison of tetrathiomolybdate and trientine in a double blind study of treatment of the neurologic presentation of Wilson’s disease. Arch Neurol 2006;63:521–7.
- Yuzbasiyan-Gurkan V, Grider A, Nostrant T, Cousins RJ, Brewer Treatment of Wilson’s disease with zinc. X. Intestinal metallothionein induction. J Lab Clin Med 1992;120:380–6.
- Sturniolo GC, Mestriner C, Irato P, Albergoni V, Longo G, D’Inca Zinc therapy increases duodenal concentrations of metallothionein and iron in Wilson’s disease patients. Am J Gastroenterol 1999;94: 334–8.
- Farinati F, Cardin R, D’Inca R, Naccarato R, Sturniolo Zinc treatment prevents lipid peroxidation and increases glutathione availability in Wilson’s disease. J Lab Clin Med 2003;141: 372–7.
- Brewer GJ, Dick RD, Yuzbasiyan-Gurkan V, Johnson V, Wang Treatment of Wilson’s disease with zinc. XIII. Therapy with zinc in presymptomatic patients from the time of diagnosis. J Lab Clinical 1994;123:849–58.
- Brewer GJ, Dick RD, Johnson VD, Brunberg JA, Kluin KJ, Fink JK. Treatment of Wilsonˇıs disease with zinc. X Long-term follow-up studies. J Lab Clin Med 1999;132:264–78.
- Yuzbasiyan-Gurkan V, Brewer GJ, Abrams GD, Main B, Giacherio Treatment of Wilson’s disease with zinc. V. Changes in serum levels of lipase, amylase, and alkaline phosphatase in patients with Wilson’s disease. J Lab Clin Med 1989;114:520–6.
- Brewer GJ, Yuzbasiyan-Gurkan V, Johnson Treatment of Wilson’s disease with zinc. IX. Response of serum lipids. J Lab Clin Med 1991;118:466–70.
- Brewer GJ, Yuzbasiyan-Gurkan V, Johnson V, Dick RD, Wang Y. Treatment of Wilson’s disease with zinc. X Interaction with other anti-copper agents. J Am Coll Nutr 1993;12:26–30.
- Askari FK, Greenson J, Dick RD, Johnson VD, Brewer Treatment of Wilson’s disease. XVIII. Initial treatment of the hepatic decompensation presentation with trientine and zinc. J Lab Clin Med 2003;142:385–90.
- Gooneratne SR, Howell JM, Gawthorne JM. An investigation of the effects of intravenous administration of thiomolybdate on copper metabolism in chronic Cu-poisoned shee Br J Nutr 1981;46:469–80.
- Brewer GJ, Hedera P, Kluin KJ, Carlson M, Askari F, Dick RB, et Treatment of Wilson disease with ammonium tetrathiomolybdate: III. Initial therapy in a total of 55 neurologically affected patients and follow-up with zinc therapy. Arch Neurol 2003;60:379–85.
- Medici V, Trevisan CP, Bigotto MA, D’Inca R, Martines D, Dal Pont E, et Adverse reaction after tetrathiomolybdate treatment for Wilson’s disease: a case report. Mov Disord 2006;21:2030–2.
- Medici V, Trevisan CP, D’Inc`a R, Barollo M, Zancan L, Fagiuoli S, et Diagnosis and management of Wilson’s disease: results of a single center experience. J Clin Gastroenterol 2006;40:936–41.
- Brewer GJ, Yuzbasiyan-Gurkan V, Dick R,Wang Y, Johnson V, Brewer Does a vegetarian diet control Wilson’s disease? J Am Coll Nutr 1993;12:527–30.
- Schumacher G, Platz KP, Mueller AR, Neuhaus R, Luck W, Langrehr JM, et Liver transplantation: treatment of choice for hepatic and neurological manifestations of Wilson’s disease. Clin Transplantation 1997;11:217–24.
- Emre S, Atillasoy EO, Ozdemir S, Schilsky M, Rathna Varma CV, Thung SN, et al. liver transplantation for Wilson’s diseas Transplantation 2001;72:1232–6.
- Guarino M, Stracciari A, D’Alessandro R, Pazzaglia P, Guarino M. No neurological improvement after liver transplantation for Wilson’s dise Acta Neurol Scand 1995;92:405–8.
- Kassam N, Witt N, Kneteman N, Bain VG. Liver transplantation for neuropsychiatric Wilson disease.Can J Gastroenterol 1998;12:65–8.
- Brewer GJ, Askari Transplant livers in Wilson’s disease for hepatic, not neurological, ndications. Liver Transpl 2000;6:662–4.
- Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli-Vergani Wilson’s Disease in children: 37-year experience and revised King’s score for liver transplantation. Liver Transpl 2005;11:441–8.
- Nazer H, Ede RJ, Mowat AP, Williams Wilson’s disease: clinical presentation and use of a prognostic index. Gut 1986;27:1377–81.
- Medici V, Mirante VG, Fassati LR, Pompili M, Forti D, Del Gaudio M, et Liver transplantation for Wilson’s disease: the burden of neuropsychiatric disorders. Liver Transpl 2005;11:1056–63.
- Devesa R, Alvarez A, de las Heras G, Ramon de Miguel J Deve Wilson’s disease treated with trientine during pregnancy. J Pediatr Gastroenterol Nutr 1995;20:102–3.
- rewer GJ, Johnson VD, Dick RD, Hedera P, Fink JK, Kluin KJ. Treatment ofWilson’s disease with zinc
- XVII. Treatment during pHepatology 2000;31:364–70.
- Kannan S. Effect of zinc treatment for Wilson’s disease on zinc concentration in breast mil J Trace Elem Exp Med 2001;14:283.
- Sternlieb Wilson’s disease and pregnancy. Hepatology 2000;31: 531–2.
- Harpey JP, Jaudon MC, Clavel JP, Galli A, Darbois Cutis laxa and low serum zinc after antenatal exposure to penicillamine. Lancet 1983;2:858.
- Pinter R, Hogge WA, McPherson Infant with severe penicillamine embryopathy born to a woman with Wilson disease. Am J Med Genet 2004;128:294–8.

