
Manuscript accepted on :September 07, 2008
Published online on: --
Plagiarism Check: Yes
Malek Taher Maghsoodlou*, Noroullah Hazeri, Sayyed Mostafa Habibi Khorassani, Forough Jalili Milani, Mohammad Amin Kazemian and Mahdi Shahraki
Department of Chemistry, The University of Sistan and Balouchestan, Zahedan (Iran).
Corresponding Author E-mail:smhabibius@ yahoo.com
Abstract
The reaction between dimethyl acetylenedicarboxylate and biological active heterocyclic NH compounds such as 3-methylpyrazole and 3,6-diboromocarbazole in the presence of triphenylphosphite at room temperature led to phosphonate ester 4a-b. The configuration of compounds 4a-b (2S*,3R*)-were determined on the basis of coupling constants emerged from the Karplus equation.
Keywords
Diastereo selectivity; Heterocyclic NH compounds; biological activity; Activated acetylenic ester
Download this article as:| Copy the following to cite this article: Maghsoodlou M. T, Hazeri N, Khorassani S. M. H, Milani F. J, Kazemian M. A, Shahraki M. Diastereoselective Synthesis of Phosphonate Ester through the Reaction Between Activated Acetylenic Ester and Heterocyclic NH Compounds with Biological Activity in the Presence of Triphenylphosphite. Biomed. Pharmacol. J.2008;1(2) |
| Copy the following to cite this URL: Maghsoodlou M. T, Hazeri N, Khorassani S. M. H, Milani F. J, Kazemian M. A, Shahraki M. Diastereoselective Synthesis of Phosphonate Ester through the Reaction Between Activated Acetylenic Ester and Heterocyclic NH Compounds with Biological Activity in the Presence of Triphenylphosphite. Biomed. Pharmacol. J.2008;1(2). Available from: http://biomedpharmajournal.org/?p=427 |
Introduction
Phosphorus-carbon bond formation1-15 is an active and important research area, as new reactions are continuously being developed for the preparation of organophosphorus compounds such as phosphinates and phosphonates.16-24 Over the last few years, the quest for the synthetic efficiency has gained remarkable importance, partly due to the need reduce waste.25 Given the increasing industrial, biological and synthetic impact of organophosphorus compounds.26-30 The successful attack by nucleophilic trivalent phosphorus on a carbon atom is facilitated when the latter is part of, or conjugated with, a carbonyl group, or when it is part of an unsaturated bond otherwise activated.27-29 There are many studies on the reaction between trivalent phosphorus nucleophiles and α, β–unsaturated carbonyl compounds in the presence of a proton source such as alcohol or phenol.31,32 Previously, the pyrazole and thiazole moieties and their derivatives have been used commercially as pharmaceuticals, pesticides and dyestuffs.33 Here we wish to report on a simple one-pot synthesis of diastereoselective phosphonate esters 4 through the reaction of biological active heterocyclic NH compounds 3 and dimethyl acetylenedicerboxylate 2 in the presence of triphenylphosphite 1
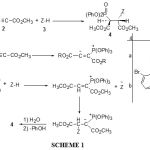 |
Scheme 1:
|
Results and Discussion
In the current work, we wish to report a simple, short time, neutral, room temperature and high diastereoselective synthesis of phosphonate esters from reaction between triphenylphosphite 1 and acetylenic ester 2 in the presence of heterocyclic NH compounds, such as 3-methylpyrazole and 3,6-diboromocarbazole 3 led to 4 in fairly high yield (see Scheme 1). These reactions were carried out in the mixture of diethyl ether and hexan (2 : 1) as solvent at room temperature and were finished within a few hours. The 1H and 13C NMR spectrum of the crude product clearly indicated the formation of phosphonate esters 4a-b. Any products other than 4a-b could not be detected by NMR spectroscopy. The structures of compounds 4a-b were confirmed by 1H, 13C, 31P NMR, mass spectrometry, IR and elemental analysis. The mass spectra of compounds 4a-b displayed molecular ion peaks at appropriate values, which were consistent with 1:1:1 adducts of heterocyclic NH compounds, DMAD and triphenylphosphite. The 300 MHz 1H NMR spectra of compound 4a displayed three sharp lines (δ= 2.23, 3.74 and 3.80) arising from methyl and methoxy protons along with signals for methine protons at δ= 4.64 ppm (1H, 2JPH=21.2 Hz, 3JHH=10.9 Hz) and δ= 5.74 ppm (1H, 3JPH=8.7 Hz, 3JHH=10.9 Hz) which appear as two doublet of doublet, respectively, for the O=P-CH-CH and O=P-CH-CH groups. The vicinal proton-proton coupling constant (3JHH) as a function of the torsion angle can be obtained from the Karplus equation.34 Typically, Jgauche varies between 1.5 and 5 Hz and Janti between 10 and 14 Hz. Observation of 3JHH=10.9 Hz for the vicinal protons in compound 4a (see Experimental section) indicates an anti arrangement for these protons. Since compound 4a possess two stereogenic center, two diastereoisomers with anti HCCH arrangements are possible. The three-bond carbon-phosphorus coupling, 3JCP, depends on configuration, as expected, transoid coupling being larger than cisoid ones. The Karplus relation can be derived from the data for organophosphorus compound with tetra and pentavalent phosphorus.35 The observation of 3JCP of 18.3 Hz for the ester C=O group (see Experimental section), is in a good agreement with the 2S,3R-4a and its mirror image 2R,3S-4a geometries (see Scheme 2). Although the presence of the 31P nucleus complicates both the 1H and 13C NMR spectra of 4a, it helps in assignment of the signals by long-range couplings with the 1H and 13C nuclei (see Experimental section). The 1H and 13C NMR spectra of 4b is similar to those of 4a, except for the ester groups, which exhibited characteristic resonances with appropriate chemical shifts (see Experimental section). The structural assignments made on the basis of the 1H and 13C NMR spectra of compounds 4a-b were supported by the IR spectra. (see Experimental section).
In conclusion, we have prepared novel diastereoselective phosphonate esters using a one-pot reaction between triphenylphosphite and dimethyl acetylenedicarboxylate in the presence of heterocyclic NH compounds such as 3-methylpyrazole and 3,6-diboromocarbazole. The present method, carries the advantage that, not only the reaction is performed under neutral conditions, but also the substances can be mixed without any activation or modifications.
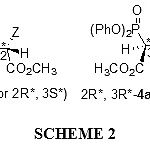 |
Scheme 2:
|
Experimental
Melting points and IR spectra of all compounds were measured on an Electrothermal 9100 apparatus and a Shimadzu IR-460 spectrometer respectively. Also, the 1H, 13C, and 31P NMR spectra were obtained from a BRUKER DRX-300 AVANCE instrument with CDCl3 as solvent at 300.1, 121.5, and 75.5 MHz respectively. In addition, the mass spectra were recorded on a Shimadzu QP 1100 EX mass spectrometer operating at an ionization potential of 70 eV. Elemental analysis for C, H and N were performed using a Heraeus CHN-O-Rapid analyzer. Dimethyl acetylenedicarboxylate, triphenlphosphite, 3-methylpyrazole and 3,6-diboromocarba- zole were purchased from Fluka, (Buchs, Switzerland) and used without further purifications.
Preparation of (2S,3R)-Dimethyl-2-(3-methylpyrazole-1-yl)-3-(diphenoxyphosphonato)-butanedioate (4a)
To a magnetically stirred solution of triphenylphosphite (0.31g, 1mmol) and 3-methylpyrazole (0.08g, 1mmol) in diethyl ether/ hexan (10ml) was added dropwise a mixture of dimethyl acetylenedicarboxylate (0.14g, 1mmol) in diethyl ether (5ml) at -10 °C over 10 min. After approximately 10 hours stirring at room temperature, the solvent was removed under reduced pressure and product washed with cold diethyl ether (2×5mL).
White powder, %93, m.p 125-127 oC, IR (KBr) (νmax, cm-1): 1740 and 1715 (C=O), 1270 (P=O). Ms, (m/z, %): 458 (M+, 3), 427 (M-OCH3, 33), 399 (M-CO2CH3, 45), 365 (M-Ph, 19), 377 (M-C4H5N2, 9), 93 (OPh, 15), 77 (Ph, 100). Anal. Calcd for C21H21N2O7P (458): C, 57.64; H, 5.02; N, 6.11, Found: C, 57.56; H, 4.95; N, 6.18. 1H NMR (300.1 MHz, δ, CDCl3): 2.23 (CH3), 3.73 and 3.80 (6H, 2s, 2OCH3), 4.64 (1H, dd, 2JPH=21.2 Hz, 3JHH=10.9 Hz, P-CH-CH), 5.74 (1H, dd, 3JPH=8.7 Hz, 3JHH=10.9 Hz, P-CH-CH), 6.05 -7.56 (13H, m, Haro). 13C NMR (75.5 MHz, δ, CDCl3): 13.60 (CH3), 47.93 (d, 1JCP=134.6 Hz, P-CH), 53.28 and 53.42 (2S, 2OCH3), 61.72 (d, 2JCP=3.5 Hz, P-C-CH), 106.06 (1C, C3H3N2), 120.16 and 120.37 (2d, 3JPC= 4.8 Hz Cortho of 2C6H5 ), 125.40 and 125.49 (Cpara of 2C6H5), 129.69 and 129.80 (Cmeta of 2C6H5), 132.98 (1C , C3H3N2), 149.70 and 150.45 (2d, 2JCP=9.6 Hz ,Cipso of 2C6H5), 150.46 (1C, C3H3N2), 166.85 (d, 2JCP=6.0 Hz, C=O), 167.74 (d, 3JCP=18.3 Hz, C=O). 31P NMR (121.5 MHz, δ, 10.38 [s, (PhO)2P(=O)].
(2S,3R)-Dimethyl-2-(3,6-diboromocarbazole-1-yl)-3-(diphenoxyphosphonato)- butanedioate (4d)
White powder, %95, m.p 163-165 oC, IR (KBr) (νmax, cm-1): 1748 and 1720 (C=O), 1271 (P=O).MS, (m/z, %): 701 (M+, 33), 642 (M-CO2CH3, 38), 608 (M-OPh, 29), 377 (M-C12H6Br2N2, 15), 77 (Ph, 100). Anal. Calcd for C25H23N2O7P (701): C, 51.36; H, 3.42; N, 1.99, Found: C, 51.49; H, 3.36; N, 2.07. 1H NMR (300.1 MHz, δ, CDCl3): 3.61 and 3.89 (6H, 2s, 2OCH3), 4.73 (1H, dd, 2JPH=21.1 Hz, 3JHH=11.8 Hz, P-CH-CH), 6.27 (1H, dd, 3JPH=7.9 Hz, 3JHH=11.8 Hz, P-CH-CH), 6.38 -8.08 (16H, m, Haro). 13C NMR (75.5 MHz, δ, CDCl3): 45.55 (d, 1JCP=136.9 Hz, P-CH), 53.56 and 53.64 (2S, 2OCH3), 55.80 (d, 2JCP=3.9 Hz, P-C-CH), 111.30, 111.99, 112.41, 113.46 and 113.66 (5C, C12H6Br2N), 119.07 and 119.77 (2d, 3JPC= 3.5 Hz Cortho of 2C6H5 ), 123.04, 123.09, 123.72. 123.85 and 125.12 (5C , C12H6Br2N), 125.40 and 125.33 (Cpara of 2C6H5), 129.40 (1C , C12H6Br2N), 129.57 and 129.60 (Cmeta of 2C6H5), 129.64 (1C , C12H6Br2N), 149.12 and 149.58 (2d, 2JCP=9.1 Hz ,Cipso of 2C6H5), 166.72 (d, 2JCP= 6.3 Hz, C=O), 168.84 (d, 3JCP=19.5 Hz, C=O). 31P NMR (121.5 MHz, δ, 9.83 [S, (PhO)2P=O].
Acknowledgements
We gratefully acknowledge financial support from the Research Council of University of Sistan and Balouchestan
References
- Maghsoodlou M. T., Habibi-Khorasani S. M., Heydari R., Hassankhani A., Marandi G., Nassiri M. and Mosaddeg E., Mol Divers, 11, 87 (2007)
- Maghsoodlou M. T., Habibi-Khorasani S. M., Niroumand U., Rostami-Charati F. and Khosrosharodi M., Phosphor, Sulfur and Silicon, 182, 647 (2007)
- Yavari I. and Karimi E., Phosphor, Sulfur and Silicon, 182, 595 (2007)
- Robiette R., Richardson J., Aggarval V. K and Harvey J. N., J. Am. Chem. Soc, 127, 13468 (2005)
- Alizadeh A. and Bijanzadeh H. R., Synthesis, 18, 3023 (2004)
- Islami M. R., Mollazehi F., Badiei A. and Sheibani H., Arkivoc, xy, 25 (2005)
- Hassani Z., Islami M. R., Sheibani H., Kalantari M. and Saidi K., Arkivoc, I, 89 (2006)
- Kalantari M., Islami M. R., Hassani Z. and Saidi K., Arkivoc, x, 55 (2006)
- Kaboudin B., Haruki T., Yamagishi T. and Yokomatsu T., Tetrahedron., 63, 8199 (2007)
- Bravo-Altamirano K., Jean-Luc. Montchamp, Tetrahedron Lett, 48, 5755 (2007)
- Benetsky E. B., Zheglov S. V., Grishina T. B., Macaev F. Z., Bet L. P., Davankov V. A. and Gavrilov K. N., Tetrahedron Lett, 48, 8326 (2007)
- Ito S., Hashino S., Morita N., Yoshifuji M., Hirose D., Takahashi M. and Kavazoe Y., Tetrahedron, 63, 10246 (2007)
- Coudray L., Abrunhosa-Thomas I. and Montchamp J. L., Tetrahedron Lett, 48, 6505 (2007)
- Morisaki Y., Ouchi Y., Naka K. and Chujo Y., Tetrahedron Lett, 48, 1451 (2007)
- Yamagishi T., Haruki T. and Yokomatsu T., Tetrahedron, 62, 9210 (2006)
- Maghsoodlou M. T., Habibi-Khorassani S. M., Rofouei M. K., Adhamdoust S. R. and Nassiri M., Arkivoc, xii, 145 (2006)
- Maghsoodlou M. T., Hazeri N., Habibi-Khorassani S. M., Saghatforoush L., Rofouei M. K. And Rezaie M., Arkivoc, xiii, 117 (2006)
- Maghsoodlou M. T., Habibi-Khorassani S. M., Heydari R. and Rostami Charati F., J.Chem. Res, 364 (2006)
- Yavari I. and Ramazani A., Phosphor, Sulfur and Silicon, 130, 73 (1997)
- Yavari I., Anary-Abbasinejad M. and Hossaini Z., Org. Biomol. Chem, 1, 560 (2003)
- Brel V. K., Synthesis, 13, 1829 (2002)
- Krawczyk Wolf W. and Sliwinski M., J. Chem. Soc. Perkin Trans 1, 1, 2794 (2002)
- Moonen K., Van. Meenen E., Verwee A. and Stevens C. V., Angewandte, 44, 7407 (2005)
- Jankowski S., Marczak J., Olczak A.and Gloka M. L., Tetrahedron Lett., 47, 3341 (2006)
- Trost M. B., Science, 254, 1471 (1991)
- Trost, M. B., Acc. Chem. Res, 35, 695 (2002)
- Hudson H. R., The Chemistry of Organophosphorus Compounds, Vol. 1. Primary, Secondary and Tertiary phosphines, Polyphosphines and heterocyclic Organophosphorus (III) Compounds, 1, pp 386-472 (Wiley, New York, 1990)
- Engel R., Synthesis of Carbon-Phosphorus Bond (CRC Press: Boca Raton, FL. 1988)
- Cadogan J. I. G., Organophosphorus Reagents in Organic Synthesis (Acade- mic Press, New York, 1979)
- Moonen K., Laureyn I. and Stevens C., Chem. Rev, 104, 6177 (2004)
- George M. V., Khetan S. K. and Gupta R. K., Adv. Heterocyclic. Chem., 19, 354 (1976)
- Burgada R., Leroux Y. and Khoshnieh Y.U., Tetrahedron Lett, 22, 3533 (1981)
- Gilchrist T. L., Heterocyclic Chemistry (Wiley, New York, 1985)
- (a) Yavari I., Islami M. R. and Bijanzadeh H. R., Tetrahedron, 55, 5547 (1999), (b) Yavari I. and Ramazani A., Phosphor, Sulfur and Silicon, 130, 73 (1997)
- Breitmaier E. and Volter W., Carbon-13 NMR Spectroscopy, VCH, pp 250 (New York, 3rd Edh., 1990)

