
Manuscript accepted on :August 14, 2008
Published online on: --
Plagiarism Check: Yes
Arinthip Thamchaipenet¹*, Surin Peyachoknagul¹, Sathaporn Jittapalapong² and Danai Pinyoowong³
¹Department of Genetics, Faculty of Science, Kasetsart University, Chatuchak, Bangkok 10900, Thailand,
²Department of Parasitology, Faculty of Veterinary Medicine, Kasetsart University, Chatuchak, Bangkok 10900, Thailand.
³Department of Small Animal and Radiology, Faculty of Veterinary Medicine, Mahanokorn University of Technology, Nongchok, Bangkok 10530, Thailand.
Abstract
Bicyclomycin resistance gene from E. canis strain Bangkok was isolated by polymerase chain reaction using primers designed from the genome sequence of E. canis strain Jake. The hypothetical Bcr protein was analysed and revealed close relationship with those of drug resistance transporter Bcr/CflA subfamily from bacteria in order Rickettsiales. Topology prediction using hidden Markov model algorithms indicated that the E. canis strain Bangkok Bcr protein was an 11-transmembrane segment protein with N-terminal out of cell and C-terminal in the cytoplasm. It contained special motifs conserved in drug transporters belonging to major facilitator superfamily (MFS). Phylogenetic analysis showed that hypothetical Bcr proteins of rickettsia species were segregated from the 12-TMS proteins and were likely to represent a new member of MFS.
Keywords
E. canis strain Bangkok; bicyclomycin; drug resistance transporter Bcr/CflA subfamily
Download this article as:| Copy the following to cite this article: Pinyoowong D, Jittapalapong J, Peyachoknagul S, Thamchaipenet A. Topology Prediction and Motifs Identification of Bicyclomycin Resistance Protein of Ehrlichia Canis Strain Bangkok. Biomed Pharmacol J 2008;1(1). |
| Copy the following to cite this URL: Pinyoowong D, Jittapalapong J, Peyachoknagul S, Thamchaipenet A. Topology Prediction and Motifs Identification of Bicyclomycin Resistance Protein of Ehrlichia Canis Strain Bangkok. Biomed Pharmacol J 2008;1(1). Available from: http://biomedpharmajournal.org/?p=175 |
Introduction
Canine monocytic ehrlichiosis (CME) is a tick-borne disease caused by Ehrlichia canis. This bacterium is obligate intracellular Gram-negative bacteria in order Rickettsiales (1). CME is an important fatal tick-borne disease that can spread worldwide and clinical data on the treatment of these are limited (2, 3, 4). E. canis was considered highly resistant organisms and drug of choice to treat CME is doxycycline (5). In Thailand CME could be considered as a serious problem for tick-transmitted infection agents (2, 6, 7).
Mavromatis et al. (8) determined a large set of proteins with transmembrane helices that associated with pathogen-host interaction from the complete genome sequences of E. canis strain Jake. One of the genes found in this genome was bicyclomycin resistance (bcr) gene, a drug resistance transporter Bcr/CflA subfamily, that involved in pathogenicity (8). Bicyclomycin is an antibiotic against a broad spectrum of Gram-negative and Gram-positive bacteria. The target of bicyclomycin is rho transcription terminating factor (9). The drug resistance transporter Bcr/CflA proteins are known as transmembrane protein efflux systems belonging to the class of the major facilitator superfamily (MFS) (10, 11, 12). Members with known activity include Bcr from Escherichia coli (13), Blt from Bacillus subtitlis (14) and Nor A from Staphylococcus aureus (15).
Transmembrane proteins are difficult to work with experimentally and very few structures have been demonstrated to date because the difficulties of expression and crystallisation (16). Alternative approaches to studying these proteins such as computational methods in particular, have been focused of much research in recent years (17, 18, 19). The topology can be predicted by methods that use physiochemical properties, such as hydrophobicity or charge (20, 21), by model-based methods, such as hidden Markov models (HMMs) (17) and neural networks (22) or using methods based on the consensus approach (23).
In this study, we cloned a gene encoding hypothetical bicyclomycin resistance protein (Bcr) from E. canis strain Bangkok. This is the first report to predict topology of the drug resistance transporter Bcr/CflA protein of E. canis. Specific motif signatures for the transmembrane protein are also proposed.
Materials and Methods
Bcr gene amplification, cloning and sequencing
DNA isolated from dog blood was prepared as previously described (7). Primers for bcr gene amplification were designed from genome sequence database of E. canis strain Jake (YP303315), namely ATT069F (5’ AGCTCATATGTGTGATATGTCTTCTGATA 3’) and ATT069R (5’ AGCTGGATCCATAATAAGCTACCCTAAGTTCC 3’). NdeI and BamHI restriction sites were added (underlined) for future expression purpose. PCR amplification was generated using both primers in a Peltier thermal cycler (MJ Research, Watertown, MA, USA), by 30 cycles of 30 s at 94.0 oC, 30 s at 58.0 oC, and 1 min at 72.0 oC, preceded by 4 min at 94.0 oC and followed by 4 min at 72.0 oC. The amplicon was purified using QIAquick PCR purification kits (QIAGEN) according to the manufacturer’s protocol. The PCR product was ligated into pGEM-T easy (Promega) and transformed into Escherichia coli DH5α. The recombinant plasmid was sequenced using both PCR primers, ATT069F and ATT069R.
Motif and topology prediction
Protein signal sequence peptide was predicted using SignalP (24). Transmembrane protein topological structures were analyzed using ConPredII (23), HMMTOP (25), SOSUI (26), TMHMM (27), and TMMOD (28). Motifs were predicted using program MAFFT (29).
Phylogenetic tree construction
Amino acid sequences comparing in this study were aligned using CLUSTALW version 1.83 XP (30). The software package PHYLIP 3.63 was used for phylogenetic analysis. The Protdist program with Jones-Taylor-Thornton matrix was used to generate a distance matrix. Seqboot, Neighbor and Consense programs were used to statistically assess the tree using bootstrap resampling. A bootstrap resampling technique of 1,000 replications were performed to statistically support the reliability of the node on the trees.
Results and Discussion
Cloning and sequencing of bcr gene from E. canis strain Bangkok
To study and identify antibiotic resistance gene from uncultured or difficult to culture bacteria such as Ehrlichia spp. are problematical worked and to date culture of Ehrlichia is limited (31). However, the technique that may use to identify these genes is polymerase chain reaction (PCR). Putative bicyclomycin resistance gene (bcr) from E. canis strain Bangkok was amplified by PCR using specific primers, ATT069F and ATT069R. The 1.1 kb PCR product was obtained (data not shown) and successfully cloned into pGEM-T easy vector. Sequence analysis showed that this gene consists of 1058 bases encoding a protein of 386 amino acids. The protein was predicted a mass of 42.45 kDa. This sequence was deposited under accession number EU880584 in the GenBank database as a new bicyclomycin resistance gene.
 |
Figure 1: The elucidation of the topology of the E. canis strain Bangkok Bcr protein. (A) Hydropathy analysis by TMHMM. The probability score for transmembrane helical regions are shown as vertical lines with the amino acid position of the ends of the predicted helices given above the bars at the top. (B) Topological model by ConPredII. Hypothetical transmembrane segments are indicated in boxes.
|
Topology prediction of the Bcr protein
Bcr amino acid sequence of E. canis strain Bangkok was analysed by BlastP and revealed sequence similarity to those of drug resistance transporter Bcr/CflA subfamily sequences from organisms in order Rickettsiales. The results indicated that Bcr amino acid sequence of E. canis strain Bangkok was very close to that of E. canis strain Jake (YP303315) with 99 % identity. It showed close relationship with 84-89 % similarity to those of genus Ehrlichia including E. chaffeensis strain Sapulpa (ZP00544976), E. ruminantium strain Welgevonden (YP180541) and E. ruminantium strain Gardel (YP19663). It shared 70 % and 56 % similarity with those of Anaplasma phagocytophilum HZ (YP504788) and Anaplasma marginale strain Maries (YP153562), respectively. Similarity values of 67-70 % were observed with those of genus Wolbachia including Wolbachia pipientis (CAQ55329), Wolbachia endosymbiont of Drosophilla willistoni (ZP01314869), Wolbachia endosymbiont of Drosophilla melanogaster (NP966176) and Wolbachia endosymbiont strain TRS of Brugia malayi (YP198034). It shared 59 % similarity with that of Neorickettsia sennetsu strain Miyayama (YP505965) and 53 % with Orientia tsutsugamushi strain Boryong (YP505965), and Orientia tsutsugamushi strain Ikeda (YP001937329).
These types of drug efflux proteins belonged to major facilitator superfamily (MSF) (10, 11, 12). Hydropathy and phylogenetic analyses of several MSF revealed that they shared a common structure and could be divided into 12 and 14 transmembrane segments (TMS) (10, 11, 12). To determine the E. canis strain Bangkok Bcr protein belonging to MFS, the amino acid sequence was first scanned for the presence of signal peptide sequences using SignalP program. It is important to identify the signal peptide prior to analysis of transmembrane type of proteins because these leader peptides that target a protein for export contain a hydrophobic region that can easily be mistaken for a transmembrane region by a prediction program (17). The result revealed that there was no such leader peptide sequence located at the N-terminal of the Bcr protein. The Bcr amino acid sequence of E. canis strain Bangkok was then predicted the hydrophobic profiles and TMS using five different computer programs; SOSUI, ConPredII, HMMTOP, TMHMM, and TMMOD. The results obtained from ConPredII, HMMTOP, TMHMM and TMMOD identified the E. canis strain Bangkok Bcr protein as an 11-TMS with N-terminal out of cell and C-terminal in the cytoplasm (Fig. 1A and B). The E. canis strain Bangkok Bcr protein contained 59.84% hydrophobic amino acids, 13 charged residues inside, 2 charged residue outside. Eleven TMSs from TMHMM were found between residues 26-48, 60-82, 86-103, 116-138, 144-166, 191-213, 228-250, 262-284, 294-316, 323-345 and 350-372 (Fig. 1A). Each position of 11 TMSs was in approximately the same positions as predicted by the four programs. In contrast, SOSUI predicted this protein as 12-TMS, with the N and C termini of the protein located in the cytoplasm. SOSUI has been originally developed to predict TM alpha-helices by using hydropathy plot model to distinguish between transmembrane and soluble proteins (18). However, HMMTOP, TMHMM and TMMOD employed hidden Markov models (HMMs) for more accurate topology prediction. Like hydropathy plot and neural network methods, they compute both sequence to structure and structure to structure relationships. The differences are that in HMMs the two steps are joining together into one integrated model (19). On the basis of the HMMs analysis we, therefore, regard the eleven membrane-span model as the most likely for the E. canis strain Bangkok Bcr protein.
 |
Figure 2: Multiple-sequence alignments of the drug resistance transporter Bcr/CflA subfamily from the order Rickettsiales. Grey background for identity 70-100 %. The consensus of motifs A, B and C were defined from conserved amino acid with an identity over 80 %. The predicted transmembrane seqments (TMS) are squared.
|
Signature motif prediction and phylogenetic relationship
Since the E. canis strain Bangkok Bcr protein was predicted as 11-TMS, all of those 13 drug resistance transporters in order Rickettsiales were then subjected to HMMTOP, TMHMM and TMMOD to analyse the transmembrane profiles. The results indicated that they were 11-TMS (data not shown). 11-TMS proteins from 14 species in order Rickettsiales were multiple aligned (Fig. 2). The sequences were scanned for the presence of MFS transporter family signature motifs A (GxLaDrxGrkxxxl), B (lxxxRxxqGxgaa) and C (gxxxGPxxGGxl) (10, 11) using the program MAFFT. Motif A was found as GPLSDxyGRrpxmL located in the cytoplasmic loop between TMS 1 and 2, motif B was found as LIxxRFiQGxGAG located within TMS 3, and motif C was conserved as SPxx[GA]P[VI]xGSxI found within TMS 4 of all 14 proteins (Fig. 2; residues in bold indicate those conserved in the MFS signature motifs). These signature motifs are typical characters of both 12-TMS and 14-TMS families of the MSF (10, 11) indicated that E. canis strain Bangkok Bcr protein and those of rickettsia species were membrane transporters belonging to MSF. However, motifs D2 and G specific for 12-TMS family, and motifs D1, H, E and F specific for 14-TMS family, could not be identified in 11-TMS family of drug transporters of rickettsias. It could, therefore, be suggested that transporters with a number of TMS smaller than 12 may be also involved in drug transport and these 14 proteins may represent a new 11-TMS family. With highly conserved sequences of motifs A, B and C has allowed us to define more precisely the sequence motif A (GPLSDxyGRrpxmL), motif B (LIxxRFiQGxGAG) and motif C (SPxx[GA]P[VI]xGSxI) specifically for the drug efflux proteins of Ehrlichia and the closely related genera Anaplasma, Neorickettsia, Orientia and Wolbachia. These signatures will be very useful for identification of a new member of the drug transporters of the 11-TMS family from other rickettsia species.
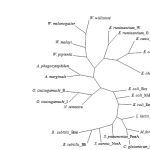 |
Figure 3: Unrooted phylogenetic tree of the drug resistance transporter Bcr/CflA subfamily from 14 rickettsia species and 10 of 12-transmembrane proteins of major facilitator superfamily (see text for details).
|
From BlastP analysis, the E. canis strain Bangkok Bcr protein showed higher similarity to those of 12-TMS family than 14-TMS family of MFS. The amino acid sequence of the E. canis strain Bangkok Bcr protein was then compared with those of 10 proteins of 12-TMS family including Bcr from Escherichia coli (X63703), Blt from Bacillus subtilis (L32599), Bmr from B. subtilis (M33768), Cmr from Corynebacterium glutamicum (U43535), EmrD from E. coli (P31442), LmrP from Lactococcus lactis (X89779), MdfA(Cmr/CmlA) from E. coli (Y08743), NorA from Staphylococcus aureus (D90119), PmrA from Streptococcus pneumoniae (AJ007367) and Tap from Mycobacterium fortuitum (AJ000283). The E. canis strain Bangkok Bcr protein shared much lower similarity (27-48 %) with these 12-TMS family than those of rickettsia species. The conserved of motifs A, B and C were also detected around N-terminal half of the proteins but lower similarity was observed at the C-terminal half (data not shown). It was previously proposed that N-terminal region of MFS proteins involved primarily in proton translocation, whereas, C-terminal half involved primarily in substrate recognition (10). An unrooted phylogenetic tree was generated from 14 drug transporters of 11-TMS of rickettsia species and 10 proteins of 12-TMS family. The members of our 11-TMS family were segregated from the 12-TMS family and grouped relatively tight in their own branch (Fig. 3). The results supported that the proposed 11-TMS family of Ehrlichia, Anaplasma, Neorickettsia, Orientia and Wolbachia were likely to represent a new family of MSF.
Acknowledgements
This research is supported by the Graduate School, Kasetsart University.
References
- Dumler J. S., Barbet A. F., Bekker C. P., Dasch G. A., Palmer G. H., Ray S. C., Rikihisa Y. and Rurangirwa F. R., Int. J. Syst. Evol. Microbiol., 51, 2145 (2001).
- Suksawat J., Pitulle C., Arraga-Alvarado C., Madrigal K., Hancock S. I. and Breitschwerdt E. B., J. Clin. Microbiol., 39, 90 (2001a).
- Rodriguez-Vivas R. I., Albornoz R. E. F. and Bolio G. M. E., Vet. Parasitol., 127, 75 (2005).
- Trapp S. M., Dagnone A. S., Vidotto O., Freire R. L., Amude A. M. and de Moris H. S. A., Vet. Parasitol., 140, 223 (2006).
- Harrus S., Waner T., Aizenberg I. and Bark H., J. Clin. Microbiol., 36, 2140 (1998).
- Suksawat J., Xuejie Y., Hancock S. I., Hegarty B. C., Nilkumhang P. and Breitschwerdt E. B., J. Vet. Intern. Med., 15, 453 (2001b).
- Pinyoowong D., Jittapalapong S., Suksawat F., Stich R. W. and Thamchaipenet A., Infect. Genet. Evol., 8, 433 (2008).
- Mavromatis K., Doyle C. K., Lykidis A., Ivanova N., Francino M. P., Chain P., Shin M., Malfatti S., Larimer F., Copeland A., Detter J. C., Land M., Richardson P. M., Yu X. J., Walker D. H., McBride J. W. and Kypides N. C., J. Bacteriol., 188, 4015 (2006).
- Kohn H. and Widger W., Curr. Drug Targets Infect. Disord., 5, 273 (2005).
- Paulsen I. T., Brown M. H. and Skurray R. A., Microbiol. Rev., 60, 575 (1996).
- Putman M., van Veen H. W. and Konings W. N., Microbiol. Mol. Biol. Rev., 64, 672 (2000).
- Paulsen I. T., Curr. Opin. Microbiol., 6, 446 (2003).
- Bentley J., Hyatt L. S., Ainley K., Parish J. H., Herbert R. B. and White G. R., Gene, 127, 117 (1993).
- Ahmed M., Lyass L., Markham P. N., Taylor S. S., Vazquez-Laslop N. and Neyfakh A. A., J. Bacteriol., 177, 3904 (1995).
- Yoshida H., Bogaki M., Nakamura S., Ubukata K. and Konno M., J. Bacteriol., 172, 6942 (1990).
- Cooper G. M. and Hausman R. E., The Cell: a Molecular Approach, 4th edn., ASM Press, Washington D.C., 820 (2007).
- Krogh A., Larsson B., von Heijne G. and Sonnhammer E. L. L., J. Mol. Biol, 305, 567 (2001).
- Arai M., Ikeda M. and Shimizu T., Gene, 304, 77 (2003).
- Punta M., Forrest L. R., Bigelow H., Kernytsky A., Liu J. and Rost B., Methods, 41, 460 (2007).
- Jayasinghe S., Hristova K. and White S. H., J. Mol. Biol., 312, 927 (2001).
- Juretic D., Zoranic L. and Zucic D., J. Chem. Inf. Comput. Sci., 42, 620 (2002).
- Rost B., Fariselli P. and Casadio R., Protein Sci., 5, 1704 (1996).
- Arai M., Mitsuke H., Ikeda M., Xia J.-X., Kikuchi T., Satake M. and Shimizu T., Nucleic. Acids Res., 32, W390 (2004).
- Bendtsen J. D., Nielsen H., von Heijne G. and Brunak S., J. Mol. Biol., 340, 783 (2004).
- Tusnady G. E. and Simon I., Bioinformatics, 17, 849 (2001).
- Hirokawa T., Boon-Chieng S. and Mitaku S., Bioinformatics, 14, 378 (1998).
- Sonnhammer E. L., von Heijne G. and Krogh A., Proc. Int. Conf. Intell. Syst. Mol. Biol., 6, 175 (1998).
- Kahsay R. Y., Gao G. and Liao L., Bioinformatics, 21, 1853 (2005).
- Katoh K., Misawa K., Kuma K. and Miyata T., Nucleic. Acids Res., 30, 3059 (2002).
- Thompson J. D., Higgins D. G., Gibson T. J., Nucleic Acids Res., 22, 4673 (1994).
- Forbes B. A., Sahm D. F. and Weissfeld A. A., J. Clin. Microbiol., 37, 2631 (1998).

