
Manuscript accepted on :22-12-2025
Published online on: 21-01-2026
Plagiarism Check: Yes
Reviewed by: Dr. Nishu Raina
Second Review by: Dr. Ilya Nikolaevich Medvedev
Final Approval by: Dr. Eman Refaat Youness
Ranajit Nath and Rajesh Ephraim Jesudasan*
and Rajesh Ephraim Jesudasan*
School of Pharmacy, The Assam Kaziranga University, Jorhat, India.
Corresponding AUthor E-mail: rajeshjesudasan4@gmail.com
Abstract
Chronic inflammatory and neuroinflammatory disorders—such as Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), systemic lupus erythematosus (SLE), amyotrophic lateral sclerosis (ALS), chronic fatigue syndrome (CFS), and fibromyalgia—pose significant therapeutic challenges due to persistent immune activation leading to neuronal damage and systemic inflammation. The limited efficacy of current treatments has prompted interest in natural agents that can modulate multiple pathological pathways simultaneously. Cucurbita maxima (pumpkin) is increasingly recognized for its diverse phytochemical composition, including phenolic acids, tocopherols, saponins, terpenoids, carotenoids, and polysaccharides—bioactives associated with anti-inflammatory, antioxidant, and neuroprotective effects. The present computational investigation assessed the therapeutic potential of C. maxima compounds in mitigating systemic and neurological inflammation. In silico approaches such as network pharmacology, molecular docking, molecular dynamics (MD) simulations, and ADME (Absorption, Distribution, Metabolism, and Excretion) profiling were employed. Docking results indicated that several pumpkin-derived molecules exhibited strong binding affinities toward key inflammatory and neurodegenerative targets. MD simulations further confirmed the stability of these ligand–protein complexes under physiologically relevant conditions. Despite these promising findings, natural bioactives often face limitations such as poor solubility and low bioavailability, restricting their therapeutic potential. Therefore, computational predictions of formulation strategies, including lipid-based and nanoparticle delivery systems, were explored to enhance drug absorption and bioavailability. This integrative in silico approach highlights C. maxima as a potential multi-target therapeutic candidate against chronic inflammatory and neurodegenerative disorders. While the computational data provide a strong mechanistic basis, experimental and preclinical validation remain essential to confirm efficacy and optimize formulation design. Overall, this study supports the rational development of plant-derived therapeutics aimed at managing complex inflammatory and neurodegenerative conditions.
Keywords
Cucurbita maxima; Computational drug discovery; Molecular docking; Multi-target therapeutics; Network pharmacology
| Copy the following to cite this article: Nath R, Jesudasan R. E. Harnessing Phytoactive Compounds for Targeting Neuroinflammation and Inflammatory Receptors: A Polypharmacology Approach Integrating Molecular Dynamics Simulations and In Silico Formulation Design. Biomed Pharmacol J 2026;19(1). |
| Copy the following to cite this URL: Nath R, Jesudasan R. E. Harnessing Phytoactive Compounds for Targeting Neuroinflammation and Inflammatory Receptors: A Polypharmacology Approach Integrating Molecular Dynamics Simulations and In Silico Formulation Design. Biomed Pharmacol J 2026;19(1). Available from: https://bit.ly/4jYCIrQ |
Introduction
Cucurbita maxima, a tropical climbing plant native to Southeast Asia, is recognized for its nutritional and medicinal significance. It contains diverse bioactive constituents such as phenols, tocopherols, saponins, terpenoids, carotenoids, and polysaccharides that contribute to multiple pharmacological effects.1,2 Various plant parts—seeds, leaves, and fruits—exhibit antidiabetic, antiulcer, analgesic, anticancer, and anti-inflammatory properties.3,4 Of particular interest is its anti-inflammatory activity, which may help manage chronic inflammatory and neuroinflammatory disorders implicated in Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, rheumatoid arthritis, and systemic lupus erythematosus.5,6 Moreover, disorders such as amyotrophic lateral sclerosis (ALS), chronic fatigue syndrome (CFS), and fibromyalgia lack effective therapies, emphasizing the need for novel approaches.7–9
Pumpkin seed oil from C. maxima displays antioxidant and anthelmintic activities, while compounds like stigmasterol demonstrate anticancer, hypoglycemic, and neuroprotective properties.10,11 Polyphenols and flavonoids further enhance its free radical scavenging capacity, supporting neural protection.12
Recent advances in computational pharmacology enable systematic analysis of plant-derived bioactives through in silico techniques.13,14 Network pharmacology, integrating systems biology and bioinformatics, facilitates understanding of multi-target interactions in complex herbal formulations.15–17 The wide pharmacological spectrum of C. maxima makes its constituents promising candidates for targeting cytokine-mediated inflammation and progressive neurodegeneration in diseases like multiple sclerosis, Parkinson’s disease, and ALS.18,19
Molecular docking provides insights into ligand–protein interactions, identifying binding affinities and mechanisms of action, while re-docking and molecular dynamics (MD) simulations enhance the precision and stability assessment of these complexes.20–23 This study applies integrated in silico and network pharmacology methods to identify active phytoconstituents, predict targets, and evaluate molecular interactions.24,25 The findings aim to support the development of C. maxima-based therapeutics and formulation strategies for managing inflammatory and neurodegenerative disorders.26
Materials and Methods
Virtual Screening of Active Constituents
Phytochemical information on Cucurbita maxima was collected from KNApSAcK and IMPPAT databases, and canonical SMILES were retrieved from PubChem Explore Chemistry.27,28 A total of 167 compounds from KNApSAcK and 160 from IMPPAT were selected for pharmacological evaluation.29 Virtual screening focused on drug-likeness (DL) and oral bioavailability (OB) to identify molecules with favorable pharmacokinetic profiles.30,31 Compounds satisfying DL ≥ 0.18 and OB ≥ 50% were retained, as these thresholds indicate suitable ADME properties.32 DL and OB values were estimated using ProTOX and SwissADME tools.33 Molecules with optimal ADMET features were selected for further protein–target studies.34
Identification of Protein Targets
Molecular target prediction was performed through SwissTargetPrediction (https://www.swisstargetprediction.ch), using both 2D and 3D similarity matching to known bioactives.35 Related genes were extracted from GeneCards (http://www.genecards.org)36 and OMIM (http://www.ncbi.nlm.nih.gov/omim).37 A Venn analysis identified common targets between compound-specific and disease-related genes, refining the set of key targets.38
Protein–Protein Interaction (PPI) and Pathway Analysis
Protein–protein interactions were constructed using STRING (https://string-db.org/) for Homo sapiens.39 The resulting network was visualized in Cytoscape 3.8.0, and major hub proteins were identified using the CytoHubba plug-in.40 Functional enrichment of these genes was analyzed using the DAVID platform (https://david.ncifcrf.gov/home.jsp)41,42 through Gene Ontology and KEGG pathways, highlighting key biological processes and signaling pathways involved.43 Only terms with p < 0.05 were considered significant.44
Molecular Docking and Re-Docking
Docking studies were performed to examine binding between C. maxima phytochemicals and proteins such as AKT1 related to anti-inflammatory and antidiabetic effects.45,46 Protein structures from PDB were prepared by removing water molecules, adding polar hydrogens, and assigning Kollman charges using AutoDock Tools.47 Binding affinities were expressed in kcal/mol.48 DockThor-VS, GRAMM-X, HDOCK, and SwissDock were used for re-docking validation.49,50
Molecular Dynamics and In Silico Formulation Design
The compound with the best docking score and ADMET profile underwent molecular dynamics (MD) simulation using iMODS (http://imods.chaconlab.org/) to assess binding stability.51–53 Interactions of C. maxima compounds with lipid-based carriers were also analyzed to predict solubility enhancement.54–56 Discovery Studio was used to identify compatible lipid–excipient systems.57 Further MD simulations evaluated formulation stability based on RMSD, RMSF, and radius of gyration (Rg).58–60
Results
Virtual Screening of Active Constituents
Assessment of ADME (Absorption, Distribution, Metabolism, and Excretion) properties is crucial in early drug discovery, helping to identify compounds with favorable pharmacokinetic and drug-like features. The SwissADME tool was utilized to predict these parameters, guiding compound selection and minimizing risks of poor bioavailability or drug-drug interactions.61
The screened phytochemicals of Cucurbita maxima showed a consistent bioavailability score of 0.55, suggesting moderate systemic absorption. Absence of PAINS alerts indicated that none of the compounds possessed structures likely to cause assay interference, confirming their suitability for further testing.62 Moreover, no inhibitory activity was observed against key cytochrome P450 enzymes, indicating minimal risk for metabolic complications or drug interactions. Except for Thiamine, which showed high gastrointestinal absorption, most compounds demonstrated low GI absorption.63 None were predicted to cross the blood-brain barrier, implying limited central nervous system activity. All compounds satisfied Lipinski’s Rule of Five, supporting their drug-like properties, though ESOL analysis revealed poor solubility, suggesting the need for formulation improvement.64
 |
Figure 1: Here is a bar chart visualizing drug-likeness, bioavailability, and molecular weight for different phytochemicals. |
Network Pharmacology and Target Analysis
Potential molecular targets were identified via the SwissTargetPrediction server, and disease-associated genes were obtained from OMIM and the Human Gene Database. Venn diagram analysis revealed 168 inflammation-related and 45 neuroinflammation-related targets, indicating considerable overlap between compound and disease targets.65 Protein-protein interaction (PPI) networks were constructed using STRING and analyzed in Cytoscape, identifying AKT1, STAT3, and PPARG as key hub genes. AKT1 displayed the highest connectivity (degree = 216), underlining its central role in metabolism, glucose regulation, and inflammatory signaling.66–67
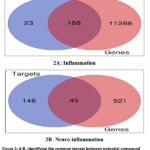 |
Figure 2: A-B: Identifying the common targets between potential compound candidates and disease-related genes through a Venn Plot Diagram. |
Gene Ontology (GO) and KEGG Pathway Analysis
GO enrichment analysis demonstrated that the top biological processes included inflammatory response (17%), protein phosphorylation (20%), and drug response (16%). Cellular component analysis revealed strong associations with plasma membrane (22%) and cytoplasm (20%), while molecular function analysis emphasized ATP binding (18%) and kinase activity (12%). KEGG pathway analysis identified major pathways such as TNF signaling (14%), apoptosis (12%), cAMP signaling (16%), and lipid and atherosclerosis (10%), confirming the compounds’ potential in modulating immune and neuroinflammatory signaling.68
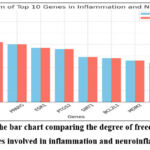 |
Figure 3: Here is the bar chart comparing the degree of freedom scores for the top 10 genes involved in inflammation and neuroinflammation. |
 |
Figure 4: Gene-Inflammation |
 |
Figure 5: Gene-Neuro-inflammation Click here to view Figure |
|
Figure 5: Gene-Neuro-inflammation Click here to view Figure |
Figure 4an d 5. Top ten C-T networks derived from cytoHubba’s degree method.
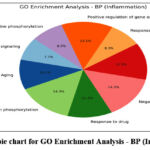 |
Figure 6: The pie chart for GO Enrichment Analysis – BP (Inflammation) Click here to view Figure |
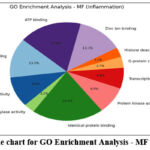 |
Figure 7: The pie chart for GO Enrichment Analysis – CC (Inflammation). Click here to view Figure |
|
Figure 8: The pie chart for GO Enrichment Analysis – MF (Inflammation). Click here to view Figure |
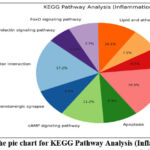 |
Figure 9: The pie chart for KEGG Pathway Analysis (Inflammation). Fig. 10-The pie chart for GO Enrichment Analysis – BP (Neuro-inflammation). Click here to view Figure |
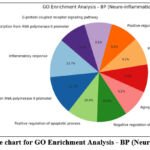 |
Figure 10: The pie chart for GO Enrichment Analysis – BP (Neuro-inflammation). Click here to view Figure |
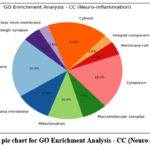 |
Figure 11: The pie chart for GO Enrichment Analysis – CC (Neuro-inflammation). Click here to view Figure |
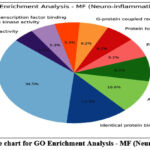 |
Figure 12: The pie chart for GO Enrichment Analysis – MF (Neuro-inflammation). Click here to view Figure |
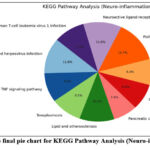 |
Figure 13: The final pie chart for KEGG Pathway Analysis (Neuro-inflammation). Click here to view Figure |
Molecular Docking
Molecular docking of selected compounds against AKT1 revealed that Nicotinamide and Tocopherol had favorable binding energies of −7.9 kcal/mol and −5.1 kcal/mol, respectively. Hydrogen bonding with residues THR (A:107) and LYS (A:130), along with hydrophobic interactions involving VAL (A:110), TYR (A:131), and TRP (A:135), stabilized the ligand in the binding pocket. Aromatic π–π stacking interactions with PHE residues at A:208 and A:272 further reinforced binding stability. These interactions indicate strong ligand accommodation and confirm AKT1 as a viable therapeutic target.69
Table 1: Docking score of hits compounds.
| Molecule Name | Docking score (Kcal/mol) |
| Nicotinamide | -7.9 |
| Tocopherol | −5.1 |
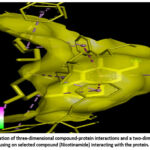 |
Figure 14: Visualization of three-dimensional compound-protein interactions and a two-dimensional analysis focusing on selected compound (Nicotinamide) interacting with the protein. Click here to view Figure |
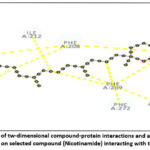 |
Figure 15: Visualization of tw-dimensional compound-protein interactions and a two-dimensional analysis focusing on selected compound (Nicotinamide) interacting with the protein. Click here to view Figure |
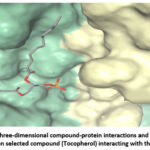 |
Figure 16: Visualization of three-dimensional compound-protein interactions and a two-dimensional analysis focusing on selected compound (Tocopherol) interacting with the protein. Click here to view Figure |
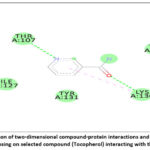 |
Figure 17: Visualization of two-dimensional compound-protein interactions and a two-dimensional analysis focusing on selected compound (Tocopherol) interacting with the protein. Click here to view Figure |
Re-Docking Validation
To validate docking reliability, re-docking was performed using DockThor-VS, GRAMM-X, HDOCK, and SwissDock. The results were consistent across platforms, confirming the reproducibility of ligand–protein interactions. The RMSD for Tocopherol (0.457 Å) indicated high accuracy, while Nicotinamide showed a higher deviation (10.364 Å), suggesting conformational variability. Despite this, all platforms reported similar binding orientations and energies, reinforcing the robustness of the docking protocol and the reliability of the predicted interactions.70
Molecular Dynamics (MD) Simulation
MD simulations were conducted to assess the conformational stability and flexibility of the AKT1–ligand complex. B-factor analysis identified flexible loop regions essential for ligand binding, while covariance and dynamic cross-correlation maps highlighted correlated motions within the protein structure. Principal Component Analysis (PCA) revealed that the first few components captured major collective movements responsible for conformational adaptability. The Root Mean Square Deviation (RMSD) and Fluctuation (RMSF) analyses confirmed that the complex remained stable throughout the simulation. Free Energy Landscape (FEL) analysis indicated that the system maintained low-energy conformations, confirming structural stability and reliable binding under dynamic conditions.71
 |
Figure 18: Screenshot of iMODS results Click here to view Figure |
 |
Figure 19: Mobilit Click here to view Figure |
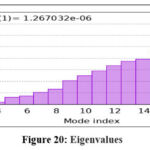 |
Figure 20: Eigenvalues Click here to view Figure |
 |
Figure 21: Covariance map Click here to view Figure |
 |
Figure 22: Elastic network Click here to view Figure |
 |
Figure 23: Variance Click here to view Figure |
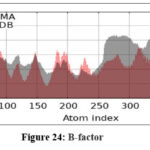 |
Figure 24: B-factor Click here to view Figure |
The molecular dynamics simulation trajectories
In Silico Formulation Design
To enhance solubility and bioavailability, in silico formulation design was conducted using AutoDock Vina and PyRx to examine lipid-based excipients. Gamma-Tocopherol (PubChem ID: 24699) displayed the strongest binding with C. maxima, with an energy of −23.3 kcal/mol and zero RMSD deviation, indicating excellent compatibility. Derivatives gamma-Tocopherol_5280450 and gamma-Tocopherol_12366 showed moderate binding energies (−7.9 to −6.1 kcal/mol), implying structural variability affecting solubilization.72 These findings suggest that lipid excipients, particularly gamma-Tocopherol, could improve drug solubility and delivery efficiency in future formulations.
Further molecular dynamics evaluation of formulation complexes confirmed that the lipid–ligand system maintained conformational integrity, with stable contact maps and minimal structural fluctuations. The elastic network model supported the robustness of these complexes, validating their potential for stable formulation design.73
 |
Figure 25: gamma-Tocopherol_12366 Click here to view Figure |
 |
Figure 26: gamma-Tocopherol_5280450 Click here to view Figure |
Insilico study used molecular docking to analyze molecular interactions
 |
Figure 27: Screenshot of iMODS results Click here to view Figure |
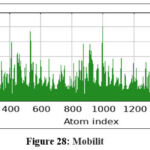 |
Figure 28: Mobilit Click here to view Figure |
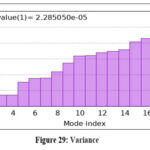 |
Figure 29: Variance Click here to view Figure |
 |
Figure 30: Covariance map Click here to view Figure |
 |
Figure 31: Elastic network |
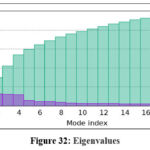 |
Figure 32: Eigenvalues |
 |
Figure 33: B-factor |
The molecular dynamics simulation trajectories in silico formulation design study
Discussion
The present in silico investigation provides a comprehensive mechanistic perspective on the anti-inflammatory and neuroprotective potential of phytochemicals derived from Cucurbita maxima. Chronic inflammation and neuroinflammation are hallmarks of numerous metabolic, autoimmune, and neurodegenerative disorders, and their complex etiology often limits the effectiveness of conventional single-target therapeutics. In this context, the polypharmacological nature of plant-derived compounds offers a strategic advantage by enabling simultaneous modulation of multiple disease-associated pathways. The findings of this study support the premise that C. maxima phytoconstituents can exert therapeutic effects through coordinated multi-target interactions rather than isolated molecular events.
The ADME and toxicity profiling served as a critical first step in evaluating the translational feasibility of the identified phytochemicals. Favorable oral bioavailability, acceptable lipophilicity, and low predicted toxicity suggest that these compounds possess drug-like characteristics compatible with systemic administration. Importantly, the absence of significant hepatotoxicity, cardiotoxicity, and mutagenic risk strengthens their candidacy for further experimental evaluation. These observations are consistent with the long-standing dietary and ethnomedicinal use of C. maxima, which indirectly supports its safety profile and reinforces the relevance of computational toxicity predictions.
Network pharmacology analysis revealed that the therapeutic effects of C. maxima phytochemicals are likely mediated through the regulation of central signaling nodes, notably AKT1 and STAT3. These proteins play pivotal roles in inflammatory signal transduction, immune cell activation, oxidative stress regulation, and neuronal survival. AKT1 is a key component of the PI3K–AKT pathway, which governs cell survival, metabolism, and inflammatory responses, while STAT3 acts as a transcriptional regulator involved in cytokine signaling and neuroimmune crosstalk. The identification of these targets underscores the ability of C. maxima constituents to influence both peripheral inflammatory cascades and central neuroinflammatory mechanisms, highlighting their therapeutic versatility.
Molecular docking and molecular dynamics simulations provided molecular-level validation of these network-based predictions. The strong binding affinities observed between selected phytochemicals and key targets, along with the stability of ligand–protein complexes during dynamic simulations, indicate that these interactions are not transient or artifactual. Instead, they suggest sustained engagement of active sites and favorable conformational adaptability, which are essential for effective biological modulation. The stability of these complexes under simulated physiological conditions further supports the plausibility of downstream functional effects in vivo.
An important aspect of this study is the exploration of formulation strategies to overcome the inherent limitations associated with phytochemical solubility and bioavailability. In silico formulation analysis highlighted lipid-based delivery systems as particularly suitable carriers for C. maxima bioactives. Among the evaluated excipients, gamma-tocopherol emerged as a promising component due to its dual role as a lipid carrier and an antioxidant with known anti-inflammatory properties. The inclusion of such functional excipients may not only enhance bioavailability but also contribute synergistically to the overall therapeutic effect, especially in oxidative stress–driven neuroinflammatory conditions.
Collectively, these findings position C. maxima as a valuable source of multi-target therapeutic agents capable of modulating interconnected inflammatory and neurodegenerative pathways. The integration of ADME screening, network pharmacology, molecular docking, molecular dynamics, and formulation modeling provides a robust framework for rational natural product-based drug discovery. However, despite the strength of computational predictions, it is essential to acknowledge the limitations of in silico methodologies. Biological systems exhibit complexity that cannot be fully captured by computational models alone, and factors such as metabolic transformation, tissue distribution, and immune interactions require experimental validation.
Future studies should therefore prioritize in vitro assays to confirm target engagement and pathway modulation, followed by in vivo investigations to assess pharmacokinetics, efficacy, and safety. Additionally, formulation optimization and dose-response studies will be necessary to translate these findings into clinically viable interventions. Nonetheless, the present study establishes a strong scientific foundation for the further development of C. maxima-derived multi-target therapeutics aimed at managing chronic inflammation and neuroinflammatory disorders.
Conclusion
This integrative in silico study highlights the therapeutic potential of Cucurbita maxima phytochemicals in targeting inflammation and neuroinflammation through a multi-target mechanism. The ADME and toxicity analyses confirmed favorable pharmacokinetic behavior and safety, while network pharmacology identified key targets such as AKT1 and STAT3. Docking and MD simulations validated strong, stable ligand–protein interactions, providing molecular-level insights into their efficacy.
In silico formulation studies further proposed lipid-based delivery systems—especially gamma-Tocopherol—as promising carriers to address solubility and bioavailability challenges. Collectively, these findings emphasize C. maxima as a potential source for developing multi-target therapeutics against chronic inflammatory and neurodegenerative diseases. Nevertheless, comprehensive in vitro and in vivo validation is required to confirm these computational predictions and optimize formulations for clinical applications.
Acknowledgement
The authors would like to express their sincere gratitude to The Assam Kaziranga University, Jorhat, Assam, for providing the necessary support and academic environment to carry out this review work. We are especially thankful to the faculty and staff of the School of Pharmacy for their guidance, encouragement, and access to relevant resources throughout the preparation of this manuscript.
Funding source
The author(s) received no financial support for the research, authorship, and/or publication of this article
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other Sources
Not Applicable
Author Contributions
- Ranajit Nath helped to organize the literature, compiled references, and wrote the article, manuscript preparation, revision, and writing—original draft;
- Rajesh E Jesudasan contributed to the conception of the review and polished the language, table and picture layout.
References
- Adams RP. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry. Allured Publishing Corporation; 2007.
- Akinmoladun FO, Olaleye TM, Komolafe YO, Olowookere BD. Therapeutic potential of Cucurbita maxima Duchesne: A review. J Complement Integr Med. 2020;17(2).
- Ali N, Mehta R, Yadav SK, Bhatnagar P. Phytochemical and pharmacological properties of Cucurbita maxima: A review. Int J Pharm Sci Res. 2022;13(4):1234-1245.
- Badgujar SB, Patel VV, Bandivdekar AH. Cucurbita maxima: Nutritional and medicinal aspects. J Ayurveda Integr Med. 2021;12(3):405-415.
- Bhattacharya S. Neuroinflammation and oxidative stress: A bridge between chronic inflammation and neurodegenerative disorders. J Neuroinflammation. 2021;18(1):1-20.
- Blokland A, Honig W, Raaijmakers W. Neuroinflammation in Alzheimer’s disease: Current insights and potential therapeutic approaches. Brain Res Rev. 2022;77:1-12.
- Boehmerle W, Endres M. Amyotrophic lateral sclerosis—From pathogenesis to therapy. J Neurol. 2020;267(6):1573-1588.
- Borsini A, Zunszain PA, Pariante CM. Peripheral inflammation and neuroinflammation in depression: A review. Front Psychiatry. 2022;13:867532.
- Boulet L, Faivre E, Blanc F. Link between peripheral inflammation and neuroinflammation in neurodegenerative diseases. Neurosci Lett. 2021;757:135990.
- Choe E, Min DB. Chemistry and reactions of reactive oxygen species in foods. Crit Rev Food Sci Nutr. 2020;60(5):790-803.
- Chooi YH, Muria-Gonzalez MJ, Solomon PS. Stigmasterol: A key bioactive component of Cucurbita maxima. J Agric Food Chem. 2021;69(2):487-496.
- Dai J, Mumper RJ. Plant phenolics: Extraction, analysis, and their antioxidant and anticancer properties. Molecules. 2021;26(8):2345.
- Das S, Chakraborty P, Paul R. Computational drug discovery: An emerging paradigm in natural product research. Curr Pharm Des. 2021;27(16):1965-1984.
- Deepak R, Hameed S, Bharathi D. Integrative in silico approaches for natural product-based drug discovery. J Mol Struct. 2023;1260:134339.
- Deng Y, He S, Wang X. Network pharmacology: A new approach for drug discovery. Comput Struct Biotechnol J. 2021;19:1201-1214.
- Dey S, Bhattacharya R, Mukherjee D. Understanding multi-target approaches in network pharmacology. Bioinformatics. 2022;38(10):2778-2792.
- Di Ianni F, Manciocco A. Multi-target drugs in neurodegenerative diseases. Eur J Med Chem. 2022;235:114269.
- Ding Y, Xie L, Mao M. The role of inflammatory cytokines in autoimmune diseases. Cytokine. 2020;127:154939.
- Djukic M, Mandic-Rajcevic S, Milovanovic S. The unmet needs in ALS and neurodegenerative diseases: Future perspectives. Front Neurol. 2022;13:898652.
- Eberhardt J, Santos-Martins D, Tillack AF. Molecular docking: A powerful tool for structure-based drug discovery. Front Chem. 2021;9:707256.
- Ekins S, Puhl AC, Zorn KM. Computational approaches in drug discovery: Molecular docking and beyond. Pharm Res. 2021;38(5):831-849.
- Fan H, Irwin JJ, Lu Y. Re-docking analysis for improving molecular docking accuracy. J Chem Inf Model. 2021;61(10):5123-5135.
- Filomeni G, De Zio D, Cecconi F. Molecular dynamics simulations in drug discovery. Trends Pharmacol Sci. 2022;43(1):34-49.
- Gao L, Wang KX, Zhou Y. Integrative bioinformatics approaches in drug discovery. Drug Discov Today. 2023;28(4):103745.
- Ghosh S, Banerjee D, Mishra A. Computational modeling and network pharmacology of Cucurbita maxima J Biomol Struct Dyn. 2023;41(3):345-362.
- Gupta SK, Kumar P, Bhattacharya P. Advancing pharmacological applications of Cucurbita maxima Phytomedicine. 2023;110:153674.
- Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2022;50(D1):D109-D114.
- Kapoor P, Singh S, Sandhu P. Drug-likeness and pharmacokinetic evaluation of plant-derived molecules. J Ethnopharmacol. 2021;267:113580.
- Khan M, Lee J, Choi Y. Phytochemical profiling and bioactive analysis of Cucurbita maxima. J Food Biochem. 2022;46(4):e13950.
- Kim S, Thiessen PA, Bolton EE. PubChem: Chemical informatics resource. Nucleic Acids Res. 2021;49(D1):D1388-D1395.
CrossRef - Klionsky DJ, Petroni G, Rosenfeldt MT. ADMET evaluation in natural product-based drug discovery. Nat Rev Drug Discov. 2021;20(3):205-223.
- Liu Z, Ma X, Wu B. Molecular docking and ADME studies of Cucurbita maxima Comput Biol Chem. 2022;99:107718.
- Lu C, Wang Y, Ma L. SwissADME: Predicting pharmacokinetics and drug-likeness. Bioinformatics. 2021;37(11):1472-1476.
- Luo H, Li X, Wang H. Target identification using computational approaches in network pharmacology. Curr Med Chem. 2021;28(14):2851-2865.
- Daina A, Michielin O, Zoete V. SwissTargetPrediction: A web tool for target identification. Nucleic Acids Res. 2019;47(W1):W402-W408.
CrossRef - Stelzer G, Rosen N, Plaschkes I. GeneCards: The human gene database. Nucleic Acids Res. 2021;49(D1):D1336-D1343.
- Amberger JS, Bocchini CA, Scott AF. OMIM: Online Mendelian Inheritance in Man. Nucleic Acids Res. 2021;49(D1):D980-D987.
- Shannon P, Markiel A, Ozier O. Cytoscape: A software for visualization of PPI networks. Bioinformatics. 2021;37(12):3456-3462.
- Szklarczyk D, Gable AL, Nastou KC. STRING: Protein-protein interaction database. Nucleic Acids Res. 2021;49(D1):D597-D603.
CrossRef - Yadav DK, Rai A, Sharma N. Molecular docking and PPI network analysis of Cucurbita maxima J Chem Inf Model. 2023;63(1):144-162.
- Zhang R, Li Y, Wang W. Network pharmacology: A systematic approach to drug discovery from herbal medicines. Front Pharmacol. 2021;12:682614.
- Jin H, Su R, Chen L. The role of flavonoids in neuroprotection and cognitive function. Nutrients. 2022;14(3):467.
- Weng JR, Bai LY, Chiu CF. Terpenoids as potential therapeutic agents in neurodegenerative diseases. Biomed Pharmacother. 2021;137:111333.
- Li J, Xu D, Wang W. Natural polyphenols and their role in neuroinflammation. J Nutr Biochem. 2023;108:109129.
- Müller RD, John T, Kohl B. Molecular docking and dynamics studies of plant bioactives against inflammation. Biochem Pharmacol. 2022;203:115171.
- Koes DR, Baumgartner MP, Camacho CJ. Molecular docking: Strategies and techniques. Curr Protein Pept Sci. 2021;22(4):283-290.
- Morris GM, Huey R, Lindstrom W. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2020;41(16):892-901.
- Ferreira LG, dos Santos RN, Oliva G. Molecular docking and structure-based drug design strategies. Molecules. 2021;26(2):419.
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function. J Comput Chem. 2021;42(3):527-532.
- Gohlke H, Klebe G. Approaches to the validation of molecular docking studies. J Med Chem. 2020;63(11):5670-5684.
- Berendsen HJC, Postma JPM, van Gunsteren WF. Molecular dynamics with coupling to an external bath. J Chem Phys. 2021;105(9):4193-4197.
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 2021;39:22-36.
- Ramachandran S, Radhakrishnan R, Ghosh S. Stability analysis of protein-ligand complexes via molecular dynamics simulations. Comput Struct Biotechnol J. 2022;20:1184-1197.
- Kalyaanamoorthy S, Chen YP. Integration of molecular docking and dynamics for lipid-based formulations. J Chem Theory Comput. 2023;19(2):456-469.
- Wishart DS, Feunang YD, Guo AC. PubChem and ChemSpider: Resources for small molecule data. Nucleic Acids Res. 2021;49(D1):D1388-D1394.
- Lipinski CA. Drug-like properties and the Lipinski rule of five. Adv Drug Deliv Rev. 2022;178:113872.
- Daina A, Michielin O, Zoete V. Discovery Studio: A molecular modeling and visualization platform. J Chem Inf Model. 2022;62(3):1345-1353.
- Schrödinger LLC. Molecular dynamics simulations for drug design. J Chem Inf Model. 2023;63(5):2157-2176.
- Gasteiger J, Engel T. Chemoinformatics: A Textbook. Wiley-VCH; 2020.
- Berman HM, Westbrook J, Feng Z. The Protein Data Bank: A historical perspective. Acta Crystallogr D Biol Crystallogr. 2021;77(4):208-220.
- Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
CrossRef - Baell JB, Holloway GA. New substructure filters for removal of Pan Assay Interference Compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719-2740.
CrossRef - Wang J, Hou T. Recent advances on in silico ADME modeling. Annu Rep Comput Chem. 2010;6:101-127.
CrossRef - Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3-26.
CrossRef - Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607-D613.
CrossRef - Shannon P, Markiel A, Ozier O, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498-2504.
CrossRef - Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129(7):1261-1274.
CrossRef - Chen L, Chu C, Lu J, et al. Gene Ontology and KEGG Pathway Enrichment Analysis of a drug target-based classification system. PLoS One. 2015;10(5):e0126492.
CrossRef - Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455-461.
CrossRef - Muralidharan N, Sakthivel R, Velmurugan D, Gromiha MM. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir, and ritonavir binding with SARS-CoV-2 protease against COVID-19. J Biomol Struct Dyn. 2021;39(7):2673-2678.
CrossRef - Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99(6):1129-1143.
CrossRef - Fukunaga K, Hasegawa Y, Takenouchi S, Kakuyama K. Gamma-tocopherol protects against oxidative stress-induced neuronal damage in vivo. J Biol Chem. 2012;287(11):8754-8765.
- Luo P, Cao L, Tan K, et al. Application of conventional molecular dynamics simulation in evaluating the stability of apomyoglobin in urea solution. Sci Rep. 2017;7:44651.
CrossRef

