
Manuscript accepted on :24-01-2026
Published online on: 19-02-2026
Plagiarism Check: Yes
Reviewed by: Dr. Baby Mittal
Second Review by: Dr. Saida Ncibi
Final Approval by: Dr. Anton R Keslav
Hasanain Abdulhameed Odhar
Department of pharmacology, College of pharmacy, Al-Zahrawi University, Karbala, Iraq
Corresponding Author E-mail:hodhar3@gmail.com
Abstract
The β-lactam antibiotics constitute a cornerstone in the management of bacterial infection. However, some strains have developed or gained resistance mechanisms to this family of antibiotics. One of the emerging resistance arsenals is the New Delhi metallo-β-lactamase-1 (NDM-1), the acquisition of this metalloenzyme can enable bacteria to deactivate the majority of β-lactam antibiotics. Despite this challenge to public health, no drug candidate was clinically approved to deactivate NDM-1 enzyme. One of the reasons for this clinical challenge is the high flexibility of NDM-1 active site. As such, it is of our interest to apply both dynamics simulation and docking tools to screen the Traditional Chinese Medicine compounds against NDM-1. The purpose of this computer-based screening is to specify a possible natural compound capable of inhibiting NDM-1 enzyme. As a result, this computational screening has identified ten potential docking hits and most of them are either triterpenes or steroids. Interestingly, some of these phytocompounds have a documented anticancer activity. Additionally, the high molecular weight for these hits is expected to limit their water solubility and pharmacokinetics profile. Then, the molecular dynamics (MD) simulation concludes that only the hit compound Solamargine is capable of maintaining a mean ligand proximity root mean square deviation (RMSD) of 3.23 Angstrom. In spite of these MD simulation results for Solamargine, the compound was unable to overcome the ligand proximity and binding energy reported to the co-crystalized ligand hydrolyzed oxacillin. One possible explanation for these superior records for the hydrolyzed oxacillin is its potential ability to form more hydrogen bonds with NDM-1 active site residues as seen in docking images. However, these in-silico findings must be further assessed against suitable bacterial strains in-vitro settings.
Keywords
Docking; Molecular dynamics simulation; NDM-1; Solamargine; TCM
| Copy the following to cite this article: Odhar H. A. Computer-based Screening of Compounds in the Traditional Chinese Medicine Database to Specify a Prospective Inhibitor of the New Delhi metallo-β-lactamase-1. Biomed Pharmacol J 2026;19(1). |
| Copy the following to cite this URL: Odhar H. A. Computer-based Screening of Compounds in the Traditional Chinese Medicine Database to Specify a Prospective Inhibitor of the New Delhi metallo-β-lactamase-1. Biomed Pharmacol J 2026;19(1). Available from: https://bit.ly/3ZGzFvh |
Introduction
The emergence of bacterial resistance to several antibiotics is deemed a remarkable health challenge nowadays. This acquired capacity of antibiotic resistance has been distributed worldwide through horizontal gene transfer among various bacterial strains.1 These strains can overcome antibiotics effect by various means like the bacterial expression of β-lactamases. The β-lactamase enzymes can catalyze the breakdown of the amide link within the β-lactam ring, resulting in the degradation of the β-lactam antibiotics.2 In this hydrolysis reaction, a nucleophilic attack is carried out against the carbonyl carbon in the β-lactam leading to ring opening and thus loss of antibiotic activity.3 Globally, about one-third of the prescribed antibiotics belong to the β-lactam class. As such, the bacterial production of β-lactamase enzymes can complicate infection treatment by boosting the mortality and morbidity rates.1,4 Based on catalysis mechanism, β-lactamases can be classified into metallo based β-lactamases (MBLs) and serine based β-lactamases (SBLs). The MBLs family utilizes divalent metal ions like zinc to perform the nucleophilic attack against β-lactam antibiotics. On the other hand, the SBLs family uses a serine amino acid in the enzyme active site pocket to attack and hydrolyze β-lactam ring. Furthermore, the β-lactamases can be divided into 4 main classes: A, B, C and D according to the enzyme amino acids sequence. It is worth mentioning that classes A, C, D of β-lactamases are considered serine-based hydrolases while class B is regarded a metalloenzyme.5 Clinically, the MBLs family is considered the most relevant to public health concerns as the metalloenzymes in this family have the ability to inactivate most of β-lactam compounds. Additionally, class B of β-lactamases can be more subdivided into B1 to B3 classes according to the alignment of common structural features.6 When considering this subclassification, the group B1 of β-lactamases looks to have the most common emergence among metalloenzymes. 7 The most eminent β-lactamase of B1 subgroup is the New Delhi metallo β-lactamase-1 (NDM-1) that has a wide spread among both clinical and environmental isolates. The NDM-1 is a potent metallo β-lactamase which was first specified in India 2009, in a Swedish tourist who was complaining of urinary tract infection.8 After that, the NDM-1 was detected in different bacterial species all over the world like Klebsiella pneumoniae and Escherichia coli. The fast and wide distribution of NDM-1 was mainly driven by the plasmid mediated transfer of the gene encoding this β-lactamase between bacterial strains.9 Usually, the NDM-1 is produced by multi-drug resistant bacteria as the plasmid piece carrying the NDM-1 encoding gene can also harbors other resistance conferring genes.10 Although some compounds were identified as NDM-1 inhibitors like Captopril, but none was clinically approved to regain the activity of β-lactam antibiotics in resistant strains. The main challenges that impeding the introduction of such clinically effective NDM-1 inhibitors are the high catalytic nature and the flexible active site of the enzyme.2,11 Structurally, the NDM-1 is consist of a single polypeptide chain with a flexible active site that contains two zinc ions to carry out the nucleophilic attack during hydrolysis reaction. These zinc ions seem to be flanked by two loops L3 and L10 that can form substrate-specific hydrophobic and hydrophilic interactions respectively. As a result, the NDM-1 active site can accommodate substrates with various molecular architectures due to the plasticity noted in loops L3 and L10.12 The place of these two loops within the NDM-1 crystal can be seen in Figure 1. Due to the essential role of NDM-1 in mediating antibiotic resistance, then there is a necessity to regain the activity of β-lactam based antibiotics through combination with an efficient NDM-1 specific inhibitor. The purpose of this in-silico study is to harness both dynamics simulation and docking tools in order to find out possible NDM-1 inhibitors by screening compounds in the Traditional Chinese Medicine (TCM) database.
 |
Figure 1: A 3D cartoon presentation for NDM-1 monomer, the N-terminus and C-terminus are colored in blue and red. The zinc ions are shown as grey spheres while loop 3 and loop 10 are both colored with magenta. |
Materials and Methods
Planning the in-silico screening study
The plan used in this structure-based in-silico screening can be summarized in three main steps: molecular docking, dynamics simulation, and prediction of chemical, pharmacokinetics and toxicity features for the best screening hits. It is worth noting that the general methodology applied in this study is the same to what we have applied in our former studies.13,14
Molecular docking
In this in-silico project, the TCM database of 2,390 compounds was virtually screened versus chain A of NDM-1 crystal (PDB: 4EYB).15 For this purpose, the online DrugRep screening site was employed for the docking of the designated database.16 This automatic screening server is using AutoDockTools (ADT) v. 1.5.6 to prepare target and ligand for the docking step,17 while the AutoDock Vina v. 1.1.2 is used by the site to perform the docking process itself.18 During this process, the employed docking grid box size was 22*22*22 Angstroms while the utilized coordinates were X: -1.0, Y: 8.0, Z: 26. As a result of this docking, the hit compounds were ranked according to their least energy of binding. From these results, just the best ten compounds were chosen for further in-silico analysis. The binding nature and orientation for each one of the best hits with a docking pose of least energy of binding was examined by both PyMOL v. 2.4.1 and Discovery studio visualizer v. 21.1.0. Additionally, the exactness of the applied docking protocol was evaluated by using the redocking method, where the co-crystalized ligand was redocked into the active location of NDM-1 monomer. Then, PyMOL v. 2.4.1 was utilized to compute the alignment degree for the conformation of both docked and co-crystalized ligands.
Computational anticipation of chemical, pharmacokinetics and toxicity properties
In this second stage of the study, several web-based sites were employed to virtually predict different chemical, pharmacokinetics, toxicity features as well as drug-likeness score for the top ten hit compounds. The employed prediction websites for this step include: SwissADME, pkCSM, ProTox v. 3.0, Molsoft L. L. C. and Cactvs v. 3.4.8.24.19–22
Molecular dynamics (MD) based simulation
The MD based simulation was used as a final step in this virtual screening project to give a better insight into the ligand binding to enzyme active position. This ligand binding potential can be predicted by allowing the ligand-target complex to move and evolve over a specified simulation duration.23 For this in-silico study, the YASARA Dynamics v. 20.12.24 tool was utilized to perform the MD simulation step.24 The detailed methodology for this simulation is the same as what we have applied in previous projects.25,26 At start, for each of the ten hits, the ligand-enzyme complex with a pose of least binding energy was submitted for 25 nanoseconds time duration. After this initial simulation, the MD results were reported as ligand proximity root mean squared deviation (RMSD). Consequently, just the hits with mean proximity to NDM-1 active site of less than 3.5 Angstrom (Å) were considered for a second extended MD based simulation for 50 nanoseconds. Again, the ligand movement as related to NDM-1 active site was measured by RMSD value while the binding energy was computed by average molecular mechanic Poisson Boltzmann surface area (MM-PBSA) approach.27 Moreover, docking complex of NDM-1 monomer and hydrolyzed oxacillin was subjected to the same simulation study and its results were used for comparative purposes.
Results
Before screening the TCM library against NDM-1 active site, the precision of the applied docking tools and parameters were validated by using the re-docking way. In this validation way, the co-crystalized ligand is taken off at first from the NDM-1 active site. Then, the extracted hydrolyzed oxacillin is docked again to the target crystal by using the same tools and parameters that were applied in the original virtual screening study. According to the redocking method, the accuracy of the applied docking step is specified by comparing the conformation of the co-crystalized ligand against the docked one. As seen in Figure 2, the conformational dissimilarity between the co-crystalized and docked hydrolyzed oxacillin was calculated as RMSD based value that was only 0.12 Å. Additionally, the energy of binding for the redocked hydrolyzed oxacillin was -8.05 Kcal/ mol.
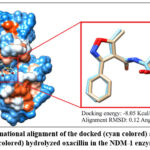 |
Figure 2: Conformational alignment of the docked (cyan colored) and co-crystalized (light brown colored) hydrolyzed oxacillin in the NDM-1 enzyme active site. |
After carrying out the structure based in-silico screening of the TCM database, the best ten hit phytocompounds with the lowest docking energy against NDM-1 were selected for more investigation. Initially, the botanical sources for these top compounds were enlisted in Table 1 together with their documented pharmacological activities. In this table, the compounds were put in order according to their calculated docking energy. As seen in Table 1, many of the listed hit compounds have an established in-vitro anticancer activity.
Table 1: A tabular review of the herbal origins and the possible pharmacological effects for the top phytocompounds generated by this structure-based computational screening.
| No. | Compound name | Herbal origin | Pharmacological effect |
| 1 | Alpha-Hederin | Hedera helix, Nigella sativa | Anticancer,28 Anti-inflammatory,29 Antioxidant 30 |
| 2 | Imperialine 3-beta-D-glucoside | Fritillaria pallidiflora, Fritillaria cirrhosa | Anticancer 31 |
| 3 | Cimiracemoside C | Cimicifuga racemosa | Antidiabetic 32 |
| 4 | Diosgenin glucoside | Tribulus terrestris | Neuroprotective,33 Anticancer,34 Antiatherosclerosis 35 |
| 5 | (25R)-Spirost-4-ene-3,12-dione | Tribulus terrestris, Persicaria chinensis | Anti-inflammatory, Antiallergic 36 |
| 6 | Dioscin | Dioscorea alata, Dioscorea opposita | Anticancer,37 Antimicrobial,38 Anti-hyperuricemia 38 |
| 7 | Actein | Cimicifuga racemose, Actaea elata | Anticancer 39 |
| 8 | Digitoxin | Digitalis purpurea | Cardiotonic,40 Anticancer 41 |
| 9 | Khasianine | Solanum nigrum | Anticancer,42 Anti-inflammatory 43 |
| 10 | Solamargine | Solanum nigrum, Solanum melongena | Anticancer 44 |
Then, several chemical characteristics were anticipated and outlined in Table 2 for these best phytocompounds. Again, the hit compounds were put in order based on their minimum docking energy. Chemically, many of these phytocompounds appear to be large molecules that are triterpenes or steroids in nature. It is clear from this table that almost all of the listed compounds have a molecular weight more than 500 g/ mol. As such, all of these compounds in Table 2 are violating Lipinski’s rule of five (RO5) 45 with the exception of hit number 5. Also, the hit number 5 in Table 2 has the least number of hydrogen bond acceptor or donor groups. Moreover, the hit number 5 is also predicted to have a polar surface area (PSA) of 52.6 Å2 which falls favorably far below the maximum allowed threshold of Veber’s rule for oral bioavailability.46 Lastly, all the compounds in Table 2 are predicted to have a logarithmic value of partition coefficient (Log P) that is below 5 and the highest value is reported for hit number 4 and 5.
Table 2: A tabular summary of the chemical features for the best phytocompounds acquired by the computer-based screening of the NDM-1 enzyme with the TCM compounds database. In this table, the listed hits were ranked according to the least docking energy.
| No. | Hit name | Chemical formula | Docking energy (Kcal/ mol) | M.W. (g/mol) | HBD | HBA | PSA (Å2) | Log P |
| 1 | Alpha-Hederin | C41H66O12 | -9.1 | 751.0 | 7 | 12 | 196 | 3.6 |
| 2 | Imperialine 3-beta-D-glucoside | C33H53NO8 | -9.1 | 591.8 | 5 | 9 | 140 | 2.3 |
| 3 | Cimiracemoside C | C35H56O9 | -8.9 | 620.8 | 5 | 9 | 138 | 3.7 |
| 4 | Diosgenin glucoside | C33H52O8 | -8.9 | 576.8 | 4 | 8 | 118 | 4.1 |
| 5 | (25R)-Spirost-4-ene-3,12-dione | C27H38O4 | -8.8 | 426.6 | 0 | 4 | 52.6 | 4 |
| 6 | Dioscin | C45H72O16 | -8.8 | 869.0 | 8 | 16 | 236 | 1.3 |
| 7 | Actein | C37H56O11 | -8.6 | 676.8 | 4 | 11 | 157 | 3.3 |
| 8 | Digitoxin | C41H64O13 | -8.5 | 764.9 | 5 | 13 | 183 | 2.3 |
| 9 | Khasianine | C39H63NO11 | -8.2 | 721.9 | 7 | 12 | 180 | 2.1 |
| 10 | Solamargine | C45H73NO15 | -8.2 | 868.1 | 9 | 16 | 238 | 1.1 |
M.W.: Molecular weight; PSA: Polar surface area; HBA: Hydrogen bond acceptor; HBD: Hydrogen bond donor; Å: Angstrom; Log P: The logarithm of partition coefficient.
Additionally, several pharmacokinetics and toxicity parameters were predicted for the best hits as presented in Table 3. The calculated drug-likeness score was also included for each of these hits in this table. As can be seen from Table 3, many of the listed TCM compounds did have a low drug-likeness score. Also, these listed hits have either a poor or moderate anticipated water solubility and this can coincide with a limited volume of distribution inside the body for these compounds as noted in this table. However, the compounds in Table 3 are expected to have a variable intestinal absorption percentage and the highest absorption was reported for hit number 5. Regarding toxicity potentials, all the ten compounds in Table 3 are speculated to be not mutagenic but some of these compounds may have a small median lethal dose (LD50) of less than 1000 mg/ Kg.
Table 3: A tabular outline of the top docking compounds’ pharmacokinetics, drug-likeness score, and toxicity features. Based on the least docking energy, the generated hit compounds were arranged in this table.
| No. | hit name | Drug-likeness | Pharmacokinetics | Toxicity | |||
| Solubility in water (mg/ml) | (%) of intestinal absorption | VDss
(L/Kg) |
AMES toxicity | LD50
(mg/ Kg) |
|||
| 1 | Alpha-Hederin | 0.84 | 2.75e-05
Poor |
38.88 | 0.07 | No | 4,000 |
| 2 | Imperialine 3-beta-D-glucoside | 0.79 | 8.66e-03
Moderate |
62.00 | 0.16 | No | 30 |
| 3 | Cimiracemoside C | -0.11 | 3.18e-04
Poor |
78.04 | 0.16 | No | 23,000 |
| 4 | Diosgenin glucoside | 0.21 | 3.17e-04
Poor |
79.65 | 0.40 | No | 8,000 |
| 5 | (25R)-Spirost-4-ene-3,12-dione | 0.23 | 7.15e-03
Moderate |
100.00 | 1.65 | No | 4,500 |
| 6 | Dioscin | 0.24 | 3.67e-04
Poor |
52.10 | 0.53 | No | 500 |
| 7 | Actein | 0.13 | 3.42e-04
Poor |
66.21 | 0.57 | No | 5,000 |
| 8 | Digitoxin | 1.05 | 3.74e-03
Moderate |
74.29 | 1.82 | No | 5 |
| 9 | Khasianine | 0.29 | 2.02e-03
Moderate |
74.85 | 0.25 | No | 500 |
| 10 | Solamargine | 0.29 | 5.79e-04
Poor |
48.10 | 0.26 | No | 100 |
VDss: The steady state-based volume of distribution; LD50: The median lethal dose.
Later, the results of MD simulation study were analyzed and summarized in Table 4 for the NDM-1 complex with each of the top hit compounds. According to the mean value of ligand movement RMSD during the first round of 25 nanoseconds simulation, only Solamargine and hydrolyzed oxacillin were able to achieve a mean proximity of less than 3.5 Å to NDM-1 active site. Therefore, these two compounds’ complexes were later subjected to a second round of 50 nanoseconds simulation time. During this extended simulation, the 10th hit Solamargine was able to keep a mean ligand proximity of 3.23 Å. On the other hand, the hydrolyzed oxacillin was capable of maintaining a mean RMSD distance of only 1.50 Å from NDM-1 active site. In the same course, the computed average MM-PBSA binding energy values were -66.33 and -54.06 Kcal/ mol for Solamargine and hydrolyzed oxacillin respectively. A thorough plot for the movement RMSD of these two compounds as a function of 50 nanoseconds simulation time duration can be observed in Figure 3.
Table 4: The MD simulation study main results for the best docking hits.
| No. | Hit name | MD simulation duration | ||
| 25 nanoseconds | 50 nanoseconds | |||
| Mean value of ligand movement RMSD (Å) | Mean value of ligand movement RMSD (Å) | Average value of MM-PBSA binding energy (Kcal/ mol) | ||
| 1 | Alpha-Hederin | 4.09 | – | – |
| 2 | Imperialine 3-beta-D-glucoside | 3.93 | – | – |
| 3 | Cimiracemoside C | 4.89 | – | – |
| 4 | Diosgenin glucoside | 4.06 | – | – |
| 5 | (25R)-Spirost-4-ene-3,12-dione | 6.18 | – | – |
| 6 | Dioscin | 3.88 | – | – |
| 7 | Actein | 3.79 | – | – |
| 8 | Digitoxin | 4.27 | – | – |
| 9 | Khasianine | 5.51 | – | – |
| 10 | Solamargine | 3.04 | 3.23 | -66.33 |
| 11 | Hydrolyzed oxacillin | 1.35 | 1.50 | -54.06 |
MD: Molecular dynamics; Å: Angstrom; RMSD: Root mean squared deviation; MM-PBSA: Molecular mechanic Poisson Boltzmann surface area.
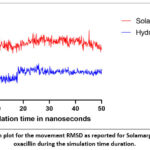 |
Figure 3: A thorough plot for the movement RMSD as reported for Solamargine and hydrolyzed oxacillin during the simulation time duration. |
Finally, evaluation of the docking 2D pictures for both hydrolyzed oxacillin and Solamargine reveals that the co-crystalized ligand (hydrolyzed oxacillin) is engaged in a higher number of hydrogen bonds with NDM-1 active location as compared to Solamargine as presented in Figure 4.
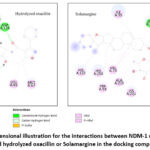 |
Figure 4: A two-dimensional illustration for the interactions between NDM-1 monomer active site and hydrolyzed oxacillin or Solamargine in the docking complex. |
Discussion
The NDM-1 is a widely distributed and a powerful metallo-β-lactamase that was detected in many clinical and environmental bacterial isolates, the acquisition of this metalloenzyme can enable bacteria to deactivate many of the β-lactam based antibiotics.9 However, the identification of effective NDM-1 inhibitors seems to be challenging due to the high plasticity of the metalloenzyme active site. This highly elastic enzyme active site can accommodate ligands with various architectures.12 Therefore, it is of interest to apply in-silico tools like docking and dynamics simulation to screen TCM library against NDM-1 active site. Thus, the objective of this computational project is to find out and introduce a possible phytocompound that can inhibit NDM-1 enzyme.
Before carrying out the virtual screening of TCM database, the exactness of the applied docking methodology was measured by using the re-docking method. As noted from Figure 2, the used docking approach appears to be precise and reliable as the conformational difference RMSD for naive versus docked hydrolyzed oxacillin was only 0.12 Å. It is well-accepted that a low RMSD value for the conformational alignment of the docked and co-crystalized ligand usually coincide with a good accuracy of the docking protocol.47
Following the structure-based computational screening step, the output hit compounds were ordered according to their least docking energy. From this screening output, only the best ten hits were chosen for more in-silico assessment. As seen in Table 1, the herbal sources and the documented activities for these hits were reported. It is very clear from this table that most of these hits are either triterpenes or steroids in nature, and many of them have in-vitro anticancer capacity. Then, several chemical features were predicted in Table 2 for these hits by using online web-based tools. As noticed from the chemical formula in this table, the hits number 2, 9 and 10 are nitrogen-based compounds known as piperidine alkaloids. Also, all the listed hits with the exception of compound 5 have a molecular weight of more than 500 g/ mol. This can be regarded as clear breach of the Lipinski’s rule of five (RO5) for oral bioavailability.45 In addition, numerous compounds in Table 2 have hydrogen bond donors or acceptors that exceed 5 or 10 groups respectively. This can be also considered another violation for the limits previously specified by RO5. Despite the fact that the compounds in Table 2 may seem to have a hydrophilic nature with a predicted logarithmic partition coefficient (Log P) of less than 5, but the high molecular weight of these compounds can restrict its water solubility. In this direction, it is easy to recognize that all the listed hits have a high anticipated polar surface area (PSA) of more than 120 Å2 with the exception of compounds 4 and 5. In fact the compound number 5 seems to have a significant low PSA value which falls in favor of Veber’s rule limits for oral bioavailability.46 Therefore, it is expected that compound 5 may have the desired chemical characteristics for good oral bioavailability. It is well-known that solubilization of a drug is a requirement for its absorption at a biological barrier. Therefore, for the hit compounds with RO5 violations, it is possible to increase water solubility by various means like reducing particles size or surfactant application.48
Next, the drug-likeness score was predicted for these herbal compounds as seen in Table 3. Based on this table, many of these hit compounds have a low drug-likeness degree but the highest score was reported for the cardiotonic drug Digitoxin. Although these best hit compounds may seem to be hydrophilic, but their high molecular weight is expected to result in a poor or moderate water solubility as noted in Table 3. Additionally, the predicted steady-state volume of distribution for these phytocompounds is anticipated to be limited due to the high molecular weight of these hits. However, the intestinal absorption percentage is believed to be variable for these compounds and the highest absorption is reported to compound 5 as noted in Table 3. Moreover, all the listed hits are expected to be non-mutagenic but the calculated median lethal dose (LD50) is reported to be less than 1000 mg/ kg for some of these compounds. As presented previously in Table 1, some of these hits are documented to have in-vitro anticancer activity and this can explain the reported low LD50 value for some of these hits. Interestingly, the least LD50 value was recorded for the drug Digitoxin which is known to have a narrow therapeutic index.49
Lastly, the binding affinity of these ten compounds was assessed by subjecting the docking complex of each hit to molecular dynamics (MD) based simulation. An outline for MD based simulation results is reported in Table 4. In this table, the results for the first round of 25 nanoseconds simulation was presented as RMSD value for mean ligand movement. It is well-accepted that MD study is a computationally intensive process,50 and in order to save both time and power we first subjected the ten hits to MD simulation for only 25 nanoseconds. Then, only those hits with a mean ligand movement RMSD of lower than 3.5 Å were chosen to another simulation for 50 nanoseconds. In this regard, it is believed that a low ligand movement RMSD value can reflect a strong binding capacity of the ligand to enzyme active location.13 Consequently, only the compounds Solamargine and hydrolyzed oxacillin were able to achieve a mean ligand proximity RMSD value of less than 3.5 Å in the first simulation as seen in Table 4. Therefore, these two compounds were then subjected to another simulation for 50 nanoseconds. As reported in Table 4, the compound hydrolyzed oxacillin was superior to Solamargine during the second simulation because it was able to achieve a lower mean ligand movement RMSD of just 1.50 Å versus 3.23 Å for Solamargine. In fact, the calculated average MM-PBSA binding energy for the hydrolyzed oxacillin was -54.06 Kcal/ mol which is better than -66.33 Kcal/ mol recorded for Solamargine. It is worth mentioning that according to the guideline for using YASARA dynamics, the more positive value of MM-PBSA binding energy usually refers to more powerful affinity of the ligand to enzyme active site.24 This closer proximity of the hydrolyzed oxacillin to NDM-1 active site can be clearly observed in Figure 3 which presents a thorough plot for ligand movement during simulation period. Further, the stronger binding affinity predicted for hydrolyzed oxacillin can be elucidated through analysis of the docking complexes as seen in Figure 4. It is obvious from this figure that the hydrolyzed oxacillin can be engaged in a higher number of hydrogen interactions with NDM-1 active location when compared to Solamargine. To sum up, it is very clear that the hydrolyzed oxacillin may be a better inhibitor to NDM-1 than does the Solamargine. As noted from MD study, the hydrolyzed oxacillin has a closer proximity to NDM-1 active site with a higher MM-PBSA binding energy. Also, the hydrolyzed oxacillin may participate in a higher number of hydrogen bonds when its docking complex was compared with that of Solamargine.
Conclusion:
In this computational project, the TCM database of compounds was screened by using dynamics-based simulation, docking and several prediction web-based tools. The aim of this structure-based computational screening is to specify a potential inhibitor of an emerging antibiotics resistance enzyme known as the New Delhi metallo-β-lactamase-1 (NDM-1). The acquisition of this hydrolyzing enzyme by plasmid mediated transfer can enable bacteria to inactivate most of the β-lactam based antibiotics. Despite the role of this metalloenzyme in driving the bacterial resistance against the β-lactam based antibiotics, the introduction of a clinically approved NDM-1 inhibitor appears to be challenging due to the high flexibility of enzyme active site. Based on the molecular docking in this study, ten phytocompounds were identified as potential NDM-1 ligands. The identified hits appear to be triterpenes or steroids with a large molecular weight. As predicted by several web-based tools, the high molecular weight of these docking hits can adversely affect the water solubility and pharmacokinetics profile for these compounds. Also, several of these hit compounds are expected to have a low median lethal dose LD50 due to their already documented in-vitro anticancer effect. Finally, the analysis of molecular dynamics (MD) based simulation results points to the output that the compound Solamargine is the only hit capable of maintaining a mean proximity RMSD of 3.23 Å away from NDM-1 active site. However, these MD simulation results reported for Solamargine seem to be inferior when compared to co-crystalized ligand hydrolyzed oxacillin. This superior behavior reported to the hydrolyzed oxacillin during simulation may be attributed to its ability to form more hydrogen bonds than Solamargine when docking images were examined.
Acknowledgment
The author would like to thanks the college of pharmacy, Al-Zahrawi University for their support of this work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Author Contributions
The sole author was responsible for the conceptualization, methodology, data collection, analysis, writing, and final approval of the manuscript.
References
- Muteeb G, Rehman M, Ali S, Al-Shahrani A, Kamal M, Ashraf G. Phage Display Technique: A Novel Medicinal Approach to Overcome An tibiotic Resistance by Using Peptide-Based Inhibitors Against β-Lactamases. Curr Drug Metab. 2017;18(2):90-95.
CrossRef - Wang T, Xu K, Zhao L, Tong R, Xiong L, Shi J. Recent research and development of NDM-1 inhibitors. Eur J Med Chem. 2021;223.
CrossRef - Salari-jazi A, Mahnam K, Sadeghi P, Damavandi MS, Faghri J. Discovery of potential inhibitors against New Delhi metallo-β-lactamase-1 from natural compounds: in silico-based methods. Sci Rep. 2021;11(1):2390.
CrossRef - Haque S, Mansor Mathkor D, Johargy AK, et al. Molecular Modeling and simulation-based identification of inhibitors against new Delhi Metallo-Lactamase 1: Implications for bacterial antibiotic resistance. J King Saud Univ – Sci. 2024;36(8):103290.
CrossRef - Bush K, Jacoby GA. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother. 2010;54(3):969-976.
CrossRef - Oelschlaeger P, Kaadan H, Dhungana R. Strategies to Name Metallo-β-Lactamases and Number Their Amino Acid Residues. Antibiot (Basel, Switzerland). 2023;12(12).
CrossRef - Garau G, García-Sáez I, Bebrone C, et al. Update of the Standard Numbering Scheme for Class B β-Lactamases. Antimicrob Agents Chemother. 2004;48(7):2347.
CrossRef - Kumarasamy KK, Toleman MA, Walsh TR, et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infect Dis. 2010;10(9):597-602.
CrossRef - Linciano P, Cendron L, Gianquinto E, Spyrakis F, Tondi D. Ten Years with New Delhi Metallo-β-lactamase-1 (NDM-1): From Structural Insights to Inhibitor Design. ACS Infect Dis. 2019;5(1):9-34.
CrossRef - Fomda BA, Khan A, Zahoor D. NDM-1 (New Delhi metallo beta lactamase-1) producing Gram-negative bacilli: Emergence & clinical implications. Indian J Med Res. 2014;140(5):672. Accessed November 12, 2025. https://pmc.ncbi.nlm.nih.gov/articles/PMC4311323/
- Ma G, Wang S, Wu K, et al. Structure-guided optimization of D-captopril for discovery of potent NDM-1 inhibitors. Bioorganic Med Chem. 2021;29.
CrossRef - King D, Strynadka N. Crystal structure of New Delhi metallo-β-lactamase reveals molecular basis for antibiotic resistance. Protein Sci. 2011;20(9):1484.
CrossRef - Odhar HA. Screening of Traditional Chinese Medicine (TCM) Compounds for their Aromatase Inhibitory Potential by Applying Molecular Docking, Dynamics Simulation and Cytotoxicity Assay. Biomed Pharmacol J. 2025;18(3):2202-2213.
CrossRef - Odhar HA, Ibrahim AA, Hashim AF. Computational Screening of Traditional Chinese Medicine (TCM) Library to Identify Potential Inhibitors of H5N1 Avian Influenza Neuraminidase: A Molecular Docking and Dynamics Simulation Study. Biomed Pharmacol J. 2025;18(2):1590-1600.
CrossRef - King DT, Worrall LJ, Gruninger R, Strynadka NCJ. New Delhi metallo-β-lactamase: structural insights into β-lactam recognition and inhibition. J Am Chem Soc. 2012;134(28):11362-11365.
CrossRef - Gan J hong, Liu J xiang, Liu Y, et al. DrugRep: an automatic virtual screening server for drug repurposing. Acta Pharmacol Sin. 2023;44(4):888-896.
CrossRef - Morris GM, Ruth H, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30(16):2785.
CrossRef - Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J Chem Inf Model. 2021;61(8):3891-3898.
CrossRef - Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7.
CrossRef - Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem. 2015;58(9):4066.
CrossRef - Banerjee P, Kemmler E, Dunkel M, Preissner R. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024;52(W1):W513-W520.
CrossRef - Ihlenfeldt WD, Takahashi Y, Abe H, Sasaki S. Computation and management of chemical properties in CACTVS: An extensible networked approach toward modularity and compatibility. J Chem Inf Comput Sci. 2002;34(1):109-116.
CrossRef - Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99(6):1129.
CrossRef - Krieger E, Vriend G. YASARA View – molecular graphics for all devices – from smartphones to workstations. Bioinformatics. 2014;30(20):2981-2982.
CrossRef - Odhar HA, Hashim AF, Humadi SS, Ahjel SW. LIGAND-BASED VIRTUAL SCREENING OF FDA-APPROVED DRUGS TO IDENTIFY NEW INHIBITORS AGAINST LACTATE DEHYDROGENASE ENZYME OF MALARIA PARASITES. Int J Appl Pharm. 2024;16(1):255-260.
CrossRef - Odhar HA, Odhar ZA, Muhi MR. Screening of Traditional Chinese Medicine Library Against Penicillin-binding Protein 2a for Methicillin-resistant Staphylococcus aureus by Molecular Docking, Dynamics Simulation and In vitro Antimicrobial Activity. Biomed Pharmacol J. 2025;18(1):823-834.
CrossRef - Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449.
CrossRef - Adamska A, Stefanowicz-Hajduk J, Renata Ochocka J. Alpha-Hederin, the Active Saponin of Nigella sativa, as an Anticancer Agent Inducing Apoptosis in the SKOV-3 Cell Line. Molecules. 2019;24(16).
CrossRef - Gepdiremen A, Mshvildadze V, Süleyman H, Elias R. Acute anti-inflammatory activity of four saponins isolated from ivy: Alpha-hederin, hederasaponin-C, hederacolchiside-E and hederacolchiside-F in carrageenan-induced rat paw edema. Phytomedicine. 2005;12(6-7):440-444.
CrossRef - Gülçin I, Mshvildadze V, Gepdiremen A, Elias R. Antioxidant activity of saponins isolated from ivy: alpha-hederin, hederasaponin-C, hederacolchiside-E and hederacolchiside-F. Planta Med. 2004;70(6):561-563.
CrossRef - Lin Q, Qu M, Patra HK, et al. Mechanistic and therapeutic study of novel anti-tumor function of natural compound imperialine for treating non-small cell lung cancer. J Ethnopharmacol. 2020;247.
CrossRef - Moser C, Vickers SP, Brammer R, Cheetham SC, Drewe J. Antidiabetic effects of the Cimicifuga racemosa extract Ze 450 in vitro and in vivo in ob/ob mice. Phytomedicine. 2014;21(11):1382-1389.
CrossRef - Wang S, Wang F, Yang H, Li R, Guo H, Hu L. Diosgenin glucoside provides neuroprotection by regulating microglial M1 polarization. Int Immunopharmacol. 2017;50:22-29.
CrossRef - Liao WL, Lin JY, Shieh JC, et al. Induction of G2/M Phase Arrest by Diosgenin via Activation of Chk1 Kinase and Cdc25C Regulatory Pathways to Promote Apoptosis in Human Breast Cancer Cells. Int J Mol Sci. 2019;21(1).
CrossRef - Lv Y cheng, Yang J, Yao F, et al. Diosgenin inhibits atherosclerosis via suppressing the MiR-19b-induced downregulation of ATP-binding cassette transporter A1. Atherosclerosis. 2015;240(1):80-89.
CrossRef - Tsai PL, Wang JP, Chang CW, Kuo SC, Lee Chao PD. Constituents and bioactive principles of Polygonum chinensis. Phytochemistry. 1998;49(6):1663-1666.
CrossRef - Si L, Xu L, Yin L, et al. Potent effects of dioscin against pancreatic cancer via miR-149-3P-mediated inhibition of the Akt1 signalling pathway. Br J Pharmacol. 2017;174(7):553-568.
CrossRef - Liu C, Wang Y, Wu C, et al. Dioscin’s antiviral effect in vitro. Virus Res. 2013;172(1-2):9-14.
CrossRef - Zhang H, Chen Y, Huang S, et al. Development of actein derivatives as potent anti-triple negative breast cancer agents. Bioorganic Med Chem Lett. 2023;89.
CrossRef - Arispe N, Diaz JC, Simakova O, Pollard HB. Heart failure drug digitoxin induces calcium uptake into cells by forming transmembrane calcium channels. Proc Natl Acad Sci U S A. 2008;105(7):2610-2615.
CrossRef - Elbaz HA, Stueckle TA, Tse W, Rojanasakul Y, Dinu CZ. Digitoxin and its analogs as novel cancer therapeutics. Exp Hematol Oncol. 2012;1(1):4.
CrossRef - Sagini MN, Klika KD, Owen RW, Berger MR. Khasianine Affects the Expression of Sugar-Sensitive Proteins in Pancreatic Cancer Cells, Which Are Altered in Data from the Rat Model and Patients. ACS Pharmacol Transl Sci. 2023;6(5):727.
CrossRef - Yang Y, Zhang Y, Chen X, Su Z, Deng Y, Zhao Q. Khasianine ameliorates psoriasis-like skin inflammation and represses TNF-α/NF-κB axis mediated transactivation of IL-17A and IL-33 in keratinocytes. J Ethnopharmacol. 2022;292.
CrossRef - Burger T, Mokoka T, Fouché G, Steenkamp P, Steenkamp V, Cordier W. Solamargine, a bioactive steroidal alkaloid isolated from Solanum aculeastrum induces non-selective cytotoxicity and P-glycoprotein inhibition. BMC Complement Altern Med. 2018;18(1):137.
CrossRef - Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3-26.
CrossRef - Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615-2623.
CrossRef - Hevener KE, Zhao W, Ball DM, et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model. 2009;49(2):444-460.
CrossRef - Savjani KT, Gajjar AK, Savjani JK. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012;2012:195727.
CrossRef - Trenti A, Zulato E, Pasqualini L, Indraccolo S, Bolego C, Trevisi L. Therapeutic concentrations of digitoxin inhibit endothelial focal adhesion kinase and angiogenesis induced by different growth factors. Br J Pharmacol. 2017;174(18):3094.
CrossRef - Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71.
CrossRef
Abbreviations List
Å: Angstrom.
LD50: Median lethal dose.
Log P: Logarithm of partition coefficient.
MBL: Metallo-β-lactamase.
MD: Molecular dynamics.
MM-PBSA: Molecular mechanics-Poisson Boltzmann surface area.
NDM-1: New Delhi metallo-β-lactamase-1.
PSA: Polar surface area.
RMSD: Root mean square deviation.
RO5: Rule of five.
SBL: Serine-β-lactamase.
TCM: Traditional Chinese Medicine.
VDss: steady state volume of distribution.

