
Manuscript accepted on :19-09-2025
Published online on: 22-10-2025
Plagiarism Check: Yes
Reviewed by: Dr. Joyeeta Bhattacharya
Second Review by: Dr. Nitish Chaudhari
Final Approval by: Dr. Kamal Upreti
Ritika Sharma1 , Hitesh Dewangan1* and Kundan Bora2
, Hitesh Dewangan1* and Kundan Bora2
1Department of Pharmacy, University Institute of Pharma Sciences, Chandigarh University, Mohali, Punjab, India
2Department of Pharmacy, University Institute of Pharma Sciences and Research, Chandigarh University, Mohali, Punjab, India
Corresponding Author E-mail:hiteshdewangan.hd@gmail.com
Abstract
Parkinson's disease (PD) is a neurodegenerative condition that worsens with time and is defined by the buildup of defective mitochondria and the selective loss of neurons that are dopaminergic. Neuronal homeostasis relies on the delicate symphony of events involving mitophagy, the selective autophagic elimination of damaged mitochondria, and mechanistic target of rapamycin (mTOR) signaling. The intricate interplay between mitophagy and mTOR pathways is discussed in this review, drawing attention to the fact that both pathways play a dual role in neuronal survival and degeneration in PD. Hyperactivation of mTOR signaling suppresses mitophagy, which can lead to mitochondrial malfunction and increased neurodegeneration, in contrast to nutrient-rich situations when mTOR signaling promotes cell development and inhibits autophagy. On the flip side, removing damaged mitochondria and avoiding oxidative stress depend on proper mitophagy activation through the PINK1/Parkin pathway. Cellular energy shortages and neuronal death can also be caused by excessive or dysregulated mitophagy. Here, we go over the molecular processes that control the interaction of mTOR complexes, AMPK, ULK1, and autophagy-related proteins, and how dysregulation of this interaction contributes to the development of PD. In order to restore mitochondrial quality control and attenuate neurodegeneration in PD, it is promising to understand this bidirectional regulation and find treatment pathways that modulate mTOR and mitophagy.
Keywords
Autophagy; Mitophagy; Mitochondrial dysfunction; mTOR signaling; Neurodegeneration; PD; PINK1/Parkin
| Copy the following to cite this article: Sharma R, Dewangan H, Bora K. Crosstalk Between mTOR Signaling and Mitophagy Pathways: A Double-Edged Sword in PD. Biomed Pharmacol J 2025;18(October Spl Edition). |
| Copy the following to cite this URL: Sharma R, Dewangan H, Bora K. Crosstalk Between mTOR Signaling and Mitophagy Pathways: A Double-Edged Sword in PD. Biomed Pharmacol J 2025;18(October Spl Edition). Available from: https://bit.ly/47dDtc5 |
Introduction
Parkinson’s disease (PD) continues to be recognized as one of the most widespread neurodegenerative illnesses. It has a substantial impact on millions of people all over the world, who are afflicted with its incapacitating symptoms and sequelae.1 Disruption of lysosome/autophagy/mitophagy processes in cells is becoming more and more linked to the complicated and multi-faceted etiology of PD. In particular, the mechanistic target of rapamycin (mTOR) plays a key role in regulating the complex cellular autophagy process, and abnormalities in mTOR have been associated with PD etiology. Many theories have surfaced that place hope in the idea that dysregulated mTOR signaling pathways may be improved or modulated in order to arrest or reverse the overall course of PD.2 This review aims to provide a thorough discussion of what is currently known about the complex role of mTOR in PD development and progression, with a specific focus on the intricate communication between mTOR signaling pathways and the different mitophagy processes.3 In addition, we will explore the known mechanisms of PD pathogenesis, namely the mTOR signaling pathways, as well as their upstream regulators and downstream mediators. To further understand how mTOR signaling contributes to the pathophysiology of PD, we really hope that these important regulators and mediators may be identified and understood. In the end, this newfound knowledge has the potential to greatly aid in the creation of treatment techniques that target the impacts and advancement of PD, giving hope to people afflicted by this difficult impairment.4 PD is a prevalent neurodegenerative disease that is clinically identified by symptoms such bradykinesia, stiffness, postural instability, and resting tremors. It is thought that the most important pathogenic process causing these symptoms is the degeneration of substantia nigra midbrain dopaminergic (mDA) neurons.5 One of the main pathogenic features of PD is the buildup of α-synuclein into filamentous intracellular inclusions called Lewy bodies. Loss of dopaminergic transmission to the motor cortex is a major consequence of the dopaminergic nigrostriatal pathway’s gradual degradation. Those who suffer from this ailment will inevitably experience motor deficiencies as a result of this degeneration, which greatly impacts their quality of life. Also, keep in mind that PD is mostly an age-related condition; one’s chances of getting PD go up substantially as they become older. As an example, around 1% of those over 60 have this disease, and that number jumps to over 4% in those over 80. Consequently, it is critical and necessary to investigate environmental and genetic variables that may hasten the natural aging process or cause cellular homeostasis dysregulation as a result of aging-induced cellular stress, both of which might lead to the onset of sporadic PD. Everyone in the scientific community agrees that a major factor in the development of PD is the malfunction of the lysosome/autophagy pathways. There may be a “double-edged sword” at work here, as autophagy and mitophagy continue to serve a “protective” purpose across a variety of PD models. The elimination of malfunctioning organelles and the mitigation of cytoplasmic aggregate buildup might be enhanced by increased autophagy and/or mitophagy. On the flip side, proteasome activity might be inhibited by overstimulation of lysosome, autophagy, or mitophagy, leading to the buildup of smaller aggregates that are considered more hazardous. So, it’s reasonable to assume that lysosome, autophagy, and mitophagy may go out of whack as PD develops.6,7
Overview of PD
The global prevalence of neurodegenerative movement disorders is highest in PD. Around 5% of people diagnosed with PD are less than 40 years old, while about 1-2% of the population over 65 suffers from the disease. Resting tremors and muscle stiffness are symptoms of increasing motor impairment. Neuroanatomically, in the CNS of PD patients, there is an increase of intraneuronal, Lewy body-like protein clumps and a depletion of dopaminergic neurons. The primary ingredient of these aggregates is α-Synuclein, and it is thought that its misfolding and aggregation are crucial in the development of PD.8
Research into the hereditary components of PD has led to the identification of fifteen genes that have a role in familial forms of the disease. When it comes to genetic mutations, the two most frequent types of inheritance are recessive and autosomal dominant. Mutations in the autosomal dominant genes (SNCA, LRRK2, and GBA) account for around 7% of hereditary cases and 1% of sporadic cases. Scientists have discovered that in highly genetically homogeneous populations, polygenic risk factors play a role in the inherited character of the vast majority of sporadic PD cases. The common term for sporadic PD, which includes environmental and genetic risk factors, is multifactorial PD.9
Mitochondrial dysfunction and degradation are becoming acknowledged as crucial factors in the etiology of PD. Increasing data indicates that Parkinsonian toxins cause oxidative damage to mitochondria, potentially disrupting the action of respiration complex I. Numerous familial PD genes have been associated with mitochondrial function. The absence of PTEN-induced kinase 1 (PINK1) or Parkin, both genes associated with PD, has been shown to hinder mitophagy and lead to the accumulation of damaged mitochondria. These findings suggest that mitochondrial dysfunction serves as a prevalent underlying mechanism in the etiology of PD.10 Fig.1
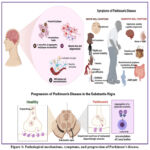 |
Figure 1: Pathological mechanisms, symptoms, and progression of Parkinson’s disease. |
The left panel illustrates key pathological hallmarks, including α-synuclein aggregation, neurofibrillary tangles, microglial activation, reactive astrocytes, and blood–brain barrier (BBB) leakage leading to neuroinflammation, ultimately resulting in neuronal loss and degeneration. The right panel depicts motor symptoms (bradykinesia, rigidity, postural instability, tremors, dystonia, walking or gait disturbances, and vocal symptoms) and non-motor symptoms (depression, anxiety, cognitive impairment, hyposmia, sweating abnormalities, melanoma, gastrointestinal disturbances, and joint pain). The lower panel shows disease progression in the substantia nigra, where healthy dopamine-producing neurons degenerate, accompanied by aggregation of α-synuclein proteins into Lewy bodies, leading to impaired dopaminergic neurotransmission.
Current Therapeutic Approaches
Comprehending the molecular mechanisms of the mitophagy pathway in PD indicates multiple opportunities for therapeutic advancement. Numerous contemporary initiatives focus on the prominent E3 ubiquitin ligase Parkin, which orchestrates the attachment of ubiquitin to proteins in the outer mitochondrial membrane.11 Ubiquitin is identified by the p62 and NBR1 ubiquitin-binding domains, which connect mitophagy to the autophagy apparatus. While Parkin is predominantly considered the focal point of PD, research into small compounds that augment the PINK1–Parkin mitophagy pathway is relatively novel. These initiatives have created a comprehensive repository of modulatory chemicals and a series of cellular assays to uncover enhancers and inhibitors of the PINK1–Parkin pathway. Additional examination of the withaferin A mechanism of action has demonstrated a comparable stimulation of 26S proteasomal degradation of Mfn2. Additional small-molecule enhancers of mitophagy that depend on mitochondrial ROS buildup include compound K and epigallocatechin gallate. Compound K is a ginsenoside derivative that promotes mitophagy through the activation of AMPK and its downstream effector, SIRT3.12
Furthermore, the newly discovered Senequin chemical operates via the small GTPase RhoB and is purported to function antagonistically to the previously recognized mitophagy activator. The significance of these findings to PD has yet to be determined. Alongside pharmaceutical improvement of mitophagy, alternative strategies targeting the mitophagy pathway are being explored. The utilization of molecular chaperones, including Drp1, PINK1, and Parkin, has been proposed to augment Parkin activity and reinstate mitophagy. This technique aimed to utilize the proteasome as a chaperone; however, much study is necessary to validate and refine it. The utilization of tiny compounds that disrupt the upstream processes of PINK1 and Parkin should aid in mitigating the detrimental effects of these genetic anomalies in monogenic PD. Alternative mitophagy-independent strategies for therapeutic advancement encompass the restoration of dysregulated lipid metabolism and the augmentation of mitochondrial biogenesis.13
This review emphasizes the potential of therapeutically altering the mitophagy system; nevertheless, it is important to note that as understanding of the pathway increases, the possibility of PD-modifying therapies beyond this channel becomes evident. Screening for small compounds that promote mitochondrial biogenesis, and mitoprotective antioxidant transcription factors is in progress. The overexpression of aggregation-prone proteins, such as α-synuclein, can impair mitochondrial morphology in cell cultures. Nonetheless, inhibiting this disturbance does not rectify ensuing deficiencies in mitophagy, indicating that mitochondrial quality control is a resilient mechanism. The presence of mitochondria with impaired components also produces systemic effects that influence the process. Independently targeting mitophagy and mitochondrial biogenesis is expected to enhance the overall efficacy of mitochondrial quality control somewhat.14
mTOR Signaling Pathway
The mechanistic target of rapamycin (mTOR) is a serine-threonine kinase including two multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The former is downstream of the insulin receptor pathway and comprises mTOR, raptor, mLST8, and PRAS40, while the latter consists of mTOR, rictor, and mLST8. Subsequent to its identification in yeast, TOR was determined to be the target of rapamycin and a crucial regulator of growth and metabolism throughout all kingdoms. The mTOR pathway amalgamates signals from growth factors, nutrition, energy levels, and stress to modulate several cellular activities, such as protein synthesis, lipid biogenesis, oxidative metabolism, and autophagy. It is thus involved in various diseases, including neurodegeneration, cancer, and metabolic disorders. Rapamycin is a natural compound, immunosuppressant, and autophagy inducer currently undergoing evaluation as a therapeutic treatment in many preclinical models, including those for neurodegeneration.15
Environmental variables such as aging, oxidative stress, mitochondrial dysfunction, and neuroinflammation are implicated in the etiology of PD. The identification of genetic variants associated with PD (mostly in familial cases, but also in certain sporadic instances) has implicated biological pathways in all key cellular functions, encompassing protein homeostasis, mitochondrial activity, neuroinflammation, and lipid homeostasis.16 Numerous proteins associated with PD, including α-synuclein, parkin, DJ-1/PARK7, PARK2, and LRRK2, are connected to, modulate, or are modulated by the mTOR pathway. Certain genetic variables associated with PD, including PINK1 and parkin, function upstream of mTOR, inhibiting its activity to promote mitophagy and preserve mitochondrial homeostasis. 6-OHDA stimulates p-beta-TrCP protein expression through oxidative stress but inhibits the upregulation of p-bet-Prx3 and p-TFEB protein expression via unidentified mechanisms.17 Nonetheless, 6-OHDA-induced mitophagy was not facilitated by p62 or rho-GTPase, and it elevated β-CATENIN protein levels and mitophagy in SK-N-SH cells. The park-independent mitophagy route was confirmed by overexpressing MAP1 LC3B to immobilize the mitophagy substrate. Moreover, iron overload replicated 6-OHDA-induced damage and mitochondrial impairments, while the mitophagy modulator DHEB mitigated PD-related damage by preserving mitochondrial homeostasis.18
Basic Mechanisms of mTOR
mTOR (mechanistic target of rapamycin) is a substantial multi-domain protein consisting of HEAT and FAT domains. HEAT-type repeats are key participants in protein-protein interactions, and mTOR exemplifies this by facilitating the building of mTORC complexes with many proteins, including Raptor, Rictor, mLST8, and Deptor.19 The phosphorylation of mTORC1 targets by mTOR induces anabolic functions and inhibits catabolic processes. Under nutrient-rich circumstances, mTORC1 regulates autophagy by phosphorylating and inhibiting essential autophagy components ULK1, ATG13, and FIP200. TBK1 is pivotal in maintaining the fidelity of phosphorylation under mTORC1-inhibited circumstances. Nonetheless, mTORC1 does not phosphorylate ULK1, ATG13, or FIP200 under hunger conditions, and the mechanism by which phosphorylation technology inhibits mTORC1 remains unclear.20 Recent research has shown that not all mTORC1 targets engage with mTORC1 through the TOR signaling (TOS) motif. Targets possessing a TOS motif undergo phosphorylation exclusively when the TSC-Rheb and Rag GTPase pathways are activated. Conversely, TEFB is susceptible to any disturbance that alters the nucleotide to which RagC is attached. The mTOR kinase inhibits autophagy when associated with mTORC2. Growth factor signaling stimulates mTORC2, which phosphorylates and activates Akt and additional members of the AGC kinase family. Targeting mTORC2 leads to the deacetylation of FoxO1, enabling its nucleus localization and subsequent activation of ATG gene transcription. The acetylation of FoxO1 in the cytoplasm facilitates the swift initiation of autophagy, even under conditions of elevated growth factor signaling. Nonetheless, mTORC2 is positioned upstream in the regulation of autophagy by SGK1, underscoring the significance of SGK1 in particular stress circumstances. The influence of mTORC1 and mTORC2 on autophagy suppression encompasses both a direct inhibition of phagophore-autophagosome formation and a prolonged restriction of the degradative machinery.21 Fig. 2
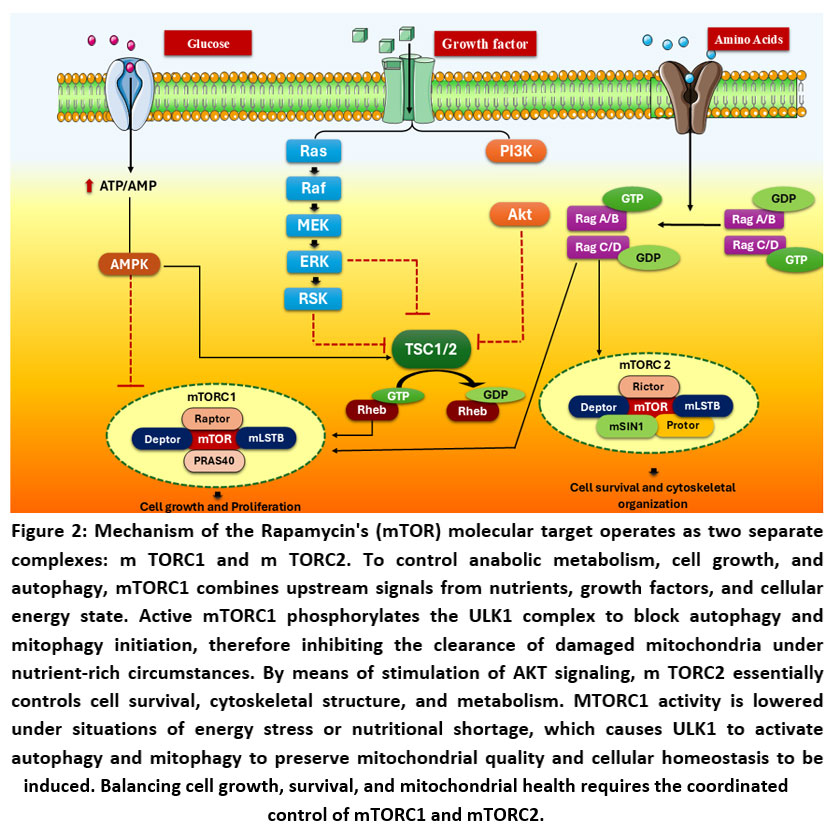 |
Figure 2: Mechanism of the Rapamycin’s (mTOR) molecular target operates as two separate complexes: m TORC1 and m TORC2. |
(AMPK: AMP-activated protein kinase, ATP: adenosine triphosphate, AMP: Adenosine monophosphate, Raf: Rapidly accelerated fibrosarcoma kinase, MEK: Mitogen-activated protein kinase kinase (MAPK/ERK kinase), ERK: Extracellular signal-regulated kinase, RSK: Ribosomal S6 kinase, PI3K: Phosphoinositide 3-kinase, Akt: Protein kinase B, TSC1/2: Tuberous sclerosis complex 1 and 2, Rheb: Ras homolog enriched in brain, mTORC1: Mechanistic target of rapamycin complex 1, mTORC2: Mechanistic target of rapamycin complex 2, mTOR: Mechanistic target of rapamycin, Raptor: Regulatory associated protein of mTOR, mLST8: Mammalian lethal with SEC13 protein 8 (also known as GβL), PRAS40: Proline-rich Akt substrate of 40 kDa, Deptor: DEP domain-containing mTOR-interacting protein, Rictor: Rapamycin-insensitive companion of mTOR, mSIN1: Mammalian stress-activated map kinase-interacting protein 1, Protor: Protein observed with Rictor)
Role of mTOR in Cellular Homeostasis
A sophisticated network of sensor proteins in eukaryotic cells precisely regulates cellular homeostasis by accurately detecting the nutritional composition of the environment, including the availability of essential elements such as amino acids, glucose, oxygen, heat, and reactive oxygen species (ROS).22 The mechanistic target of rapamycin, known as the mTOR signalling pathway, is central to this complex network and serves as a primary regulator of several homeostatic systems. mTOR is a serine/threonine kinase that assembles two vital mTORC1 and mTORC2, both of which are crucial for cellular control. Mice with a loss of the mTOR gene in neural progenitor cells exhibit substantial deficits in cortical and hippocampal neurogenesis, alongside a pronounced elevation in cell mortality rates. In the postnatal brain, the reduction of mTOR in particular neuronal subpopulations leads to gradual neuronal degeneration, which is associated with age-related cognitive deficits. Lower levels of mTOR signalling have been associated with enhanced longevity across many model organisms. Consequently, it is unsurprising that disruptions in mTOR signalling correlate with numerous age-related ailments, including as neurological diseases, different cancers, and metabolic disorders.23 The mTOR pathway is the most well investigated signalling cascade related to cellular homeostasis, successfully coordinating growth and catabolic processes. Nutrients are a crucial element of metabolic balance, and the mTOR pathway significantly mediates cellular responses to variations in nutrient levels. Any disturbance that causes diminished protein synthesis and lowered ATP generation may precipitate premature cellular senescence. Conversely, situations marked by excessive dietary intake tend to disrupt the mTOR pathway from a state of homeostasis to pathological conditions; this shift fosters growth and proliferation at the cost of critical qualities such as plasticity, stability, and cellular survival.24 This transition to a pathological state intensifies the aging process, resulting in a broad array of dysfunctions, including metabolic disorders, neurological diseases, cardiovascular degeneration, and numerous cancers. In recent years, intriguing candidate pharmaceuticals like rapamycin have attracted interest as potential therapeutic agents. The mTOR signaling pathway and its associated components play a crucial role in the regulation of cellular homeostasis, offering insights into the complex regulatory mechanisms and interactions of metabolic pathways that significantly influence cell fate.25
mTOR and Neurodegeneration
Autophagy is a vital and highly conserved cellular mechanism by which cells digest and recycle diverse proteins and defective organelles. This process entails the creation of specialized, double-membraned structures called autophagosomes, which enclose certain cellular components and then merge with lysosomes. This union commences the degradation process, enabling the elimination of damaged or superfluous cellular components. The preliminary stages of autophagosome formation are coordinated by numerous essential proteins, including Ulk1/2, Atg13, and FIP200, which collaborate to commence this vital cellular process.26 In mammals, the nutrient-sensing complex mTORC1 is crucial for controlling autophagy, especially in nutrient-rich environments, where it exerts inhibitory effects. The activation of mTORC1 specifically results in the phosphorylation of Atg1, or ULK1, which inhibits the complex’s interaction with Atg13. mTORC1 activity designates it as a principal negative regulator of autophagy.27 Phosphorylations mediated by mTORC1 efficiently obstruct the assembly of the ULK1/2 complex with Atg13, hence inhibiting the creation of autophagosomes essential for the progression of autophagy. The ULK1/2 complex is essential for maintaining equilibrium between cellular energy supply and demand by modulating the development of phagophores, which are precursors of autophagosomes. The phosphorylation statuses of ULK1/2 at several locations alter the regulation of phagophore production. Phosphorylated ULK1 (p-ULK1) significantly augments the autophagy process by phosphorylating the protein Bcl-2, which in turn boosts the activity of Beclin 1, hence increasing its ability to generate Phosphatidylinositol 3-phosphate (PI3P).28 The synthesis of PI3P is crucial for the recruitment of additional proteins necessary for the elongation and closure of phagophores. Moreover, the ULK1/2 complex selectively targets phagophores rather than other cellular membranes by engaging with specific machinery that envelops the phagophores. In times of food scarcity or starvation, the suppression of mTORC1 activates autophagy by enhancing the formation of the Atg1/ULK1 complex, so initiating the autophagic process.29 Furthermore, autophagy receptors recognize and attach to misfolded proteins or impaired organelles, promoting their destruction via this mechanism. These receptors possess ubiquitin-like domains that facilitate their interaction with ubiquitin, a tiny protein that marks substrates for destruction. Significantly, mTORC1 exerts a positive regulatory influence on Muk1, a GTPase that disrupts the interface between autophagosomes and Golgi membranes, suggesting a complex regulatory network involving mTORC1. The suppression of mTORC1 enhances Muk1 activity, hence facilitating macroautophagic clearance mechanisms that target proteasomes, ensuring effective destruction of cellular debris and the maintenance of cellular homeostasis.30
Dementia and Alzheimer’s disease (AD) are associated with mTORC1 signalling. PINK1, LRRK2, and DJ-1 are PD kinases that modulate mTOR signalling. Damage-inducing pathways linked to neurodegeneration are stoked by PAINS, whose clearance deficiencies cause neurodegeneration, by activating mTORC1 and blocking autophagy. In AD, the autophagy regulators Beclin 1 and Atg4D are suppressed.31 These species are cleared when autophagy is activated by inhibiting mTORC1. In AD, where Aβ plaques are an early sign of pathology, these results suggest that nTAs or their antecedents play a role in abnormal mTORC1 activation and flawed degradation. However, targeting mTORC1 can enhance the clearance of misfolded proteins in neurodegenerative diseases. These actions disinhibit ULK-1/2 complex formation, which promotes Atg transcription and recruitment to the phagophore mechanistically.32
Mitophagy: Mechanisms and Importance
Mitophagy is a selective breakdown process carried out by cellular autophagy mechanisms, responsible for eliminating damaged mitochondria, hence maintaining the quality and functionality of these vital organelles. This process is essential for managing energy homeostasis and preventing the neurodegeneration of dopaminergic neurons, which are critical for motor control and cognitive functioning.33 Mitochondria, the cellular powerhouses, can be eradicated via autophagy in two unique manners: selectively and non-selectively. Non-selective degradation targets several dysfunctional organelles, including the endoplasmic reticulum and peroxisomes, which may have lost their appropriate functionality.34 Conversely, selective autophagy targets specific payloads recognized by distinct receptors, ensuring that only certain organelles are digested, hence preserving cellular integrity and function. Mammalian cells have developed a complex two-step paradigm for mitophagy to selectively target and eliminate faulty mitochondria.35 The PINK1/Parkin-mediated pathway has emerged as the most extensively characterized mechanism regulating this process within this model. PINK1 is a kinase situated on the outer mitochondrial membrane, which is typically destroyed swiftly by m-AAA protease and PARL protease found in healthy mitochondria. However, when mitochondria are compromised, a critical event transpires: the mitochondrial membrane may rupture. This collapse inhibits the import of PINK1 into the mitochondria, resulting in its accumulation and stabilization on the outer mitochondrial membrane.17 The stabilization of PINK1 is essential as it subsequently recruits Parkin, an E3 ligase found in the cytosol, to the mitochondria. Upon recruitment, Parkin is phosphorylated, allowing it to promote the ubiquitination of proteins located on the mitochondrial outer membrane. This ubiquitination serves as a vital signal that recruits important selective mitophagy receptors, including p62/SQSTM1, NBR1, and NDP52, which are necessary for the identification of dysfunctional mitochondria. Subsequent to this recruitment, the mitophagy process persists as the cellular membrane may stretch and participate in the engulfment of the targeted mitochondria, ultimately resulting in the formation of a mitophagosome.36 At this point, MITOL/ARIH1, an additional E3 ubiquitin ligase, is engaged. This protein deubiquitinates ubiquitinated proteins on the outer mitochondrial membrane of the mitophagosome, thereby finalizing the regulatory components of the mitophagy pathway. Investigations using fibroblasts from Parkin or PINK1 deficient animals demonstrate that these cells had a compromised mitophagy pathway, characterized by enlarged mitochondrial aggregates that signify an inability to adequately remove malfunctioning organelles. This deficiency can substantially affect cellular health and general metabolic function.37
It is proposed that the receptor-mediated system reacts to moderate stress. Here, ROS-induced cardiolipins are shown on the outer mitochondrial membrane, which brings in Nix/BNIP3L, which fights with BCL2 to prevent mitophagosome-lysosome fusion.38 Lysosomes and damaged mitochondria can extend and form a soft link to initiate mitophagy when microtubules are present. Mitophagy could be triggered by additional pathways as well. As an example, cardiolipin functions as a sterol-transporting molecule, bringing a huge pool of cholesterol from the outer mitochondrial membrane to facilitate mitophagy, when bound to the mitochondrial membrane in the presence of phosphatidic acids. Important for maintaining mitophagy in cells, mitochondrial fragmentation and expansion could be triggered by a variety of stresses.39
Process of Mitophagy
Mitophagy refers to the selective autophagy of impaired mitochondria. Initially identified in yeast, various pathways have been established to initiate mitophagy, including those reliant on PINK1 and Parkin, two genes whose alterations lead to familial forms of PD. The activation of the PINK1-Parkin pathway entails the fast buildup of PINK1 at impaired mitochondria. PINK1 initiates cellular signaling pathways that facilitate the translocation of Parkin to the outer mitochondrial membrane. Parkin ubiquitinates substrate proteins within and next to the mitochondria, so commencing the process of mitophagy. The PINK1-Parkin pathway has garnered significant interest in the domains of autophagy and neurodegenerative diseases since its introduction.10
Mitophagy is a cellular mechanism that marks superfluous mitochondria for degradation through an autophagy-related pathway. This process is associated with various physiological and cellular functions, including cell migration, differentiation, survival, apoptosis, and diseases such as cancer, neurodegenerative disorders, and pathogenic infections.40 Mitochondria are vital cellular organelles that execute critical duties and possess their own genetic material, which is separate from the nuclear genome. Mitochondria execute other additional roles, such as calcium homeostasis, heme production, the biogenesis and turnover of metabolic enzymes, and iron metabolism. A dysfunctional mitochondrion can result in the accumulation of mitochondrial DNA mutations that adversely affect cellular function and viability, thereby being implicated in numerous neurodegenerative diseases, including AD, Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and PD.41 Numerous modifications to mitophagy have been identified in several neurodegenerative illnesses, encompassing defects, upregulation, and downregulation of mitophagy processes, along with impairments in the conventional PINK1/Parkin pathway. Comprehending the processes involved in mitophagy will elucidate the potential mechanisms altered in neurodegenerative diseases, and the disease-specific alterations may reveal novel therapeutic targets that address the defects while avoiding non-specific cellular consequences of rectifying all defects.42
Mitophagy is a crucial process in maintaining mitochondrial homeostasis; it is the process of selective breakdown of mitochondria, which begins with their encapsulation in autophagosomes and continues in the lysosome. Because the process of autophagosome production is essential for the maintenance of healthy neurons, impaired mitophagy has been linked to accelerated neurodegeneration and PD. The mitophagy pathways that are not dependent on Parkin are known as Parkin-independent mitophagy, and they mainly include receptor-mediated, lipid-mediated, and ubiquitin-mediated mitophagy, all of which play important roles in the development of PD.43
Parkin/PINK1-mediated mitophagy
In cases with recessive PD, the underlying genetic etiology is usually a mutation in either the serine/threonine kinase PINK1 or the E3 ubiquitin ligase Parkin. Mitophagy mediated by Parkin and PINK1 is a complex process. At basal circumstances, mitochondrial protease cleaves PINK1 at its destination, the inner mitochondrial membrane (IMM). Then, an N-end rule route carries the shortened PINK1 to the proteasome, where it will be degraded. When mitochondrial membrane potential collapses due to stress or injury, PINK1 is unable to translocate but instead accumulates on the outer mitochondrial membrane (OMM) and is autophosphorylated. At the Serine 65 phosphorylation site, PINK1 initiates the recruitment of Parkin from the cytosol to the mitochondria by phosphorylating OMM-conjugated ubiquitins.44 On mitochondria, when Parkin is also activated by PINK1 phosphorylation, phosphorylated ubiquitin may serve as an anchor for Parkin in addition to activating it. Phosphorylation of Parkin by ubiquitin initiates a cascade of structural changes that transform the enzyme from a quiescent state with a conformation that inhibits itself to an active, promiscuous E3 ligase that non-selectively facilitates the ubiquitination of OMM proteins.45 To prepare mitochondria for autophagic clearance, Parkin ubiquitinates and degrades Miro, one of its substrates. This arrests mitochondrial mobility. In order to speed up the mitophagy cascade, PINK1 can phosphorylate ubiquitin that is linked to the substrates of injured mitochondria. This, in turn, recruits and activates additional Parkin.10 Autophagy receptors, such as OPTN and NDP5223, then identify the ubiquitinated substrates as a breakdown signal. This happens simultaneously with the activation of TBK1 by Parkin/PINK1. TBK1 phosphorylates and attracts OPTN and NDP52 to the depolarized mitochondria, where they anchor to the LC3-coated autophagosome that is just beginning to develop. The procedure is carried out by means of a positive feedback loop that relies on ATG8 and involves the formation of an isolation membrane enclosing the mitochondria. After engulfing the injured mitochondria, the autophagosome merges with the lysosome to release acidic hydrolases. When autophagosome lysosome fusion causes mitochondria to degrade, mitophagy is complete. Also, we’ll talk about certain Parkin-independent mitophagy routes later; these might be used to make up for the failure of Parkin-mediated mitophagy.46
Genetic and pathological evidence of Parkin/PINK1-mediated mitophagy
When PINK1 and Parkin are mutated, they no longer perform their physiological roles in mitophagy. Not only has the molecular mechanism behind the Parkin/PINK1 pathway been elucidated by extensive investigations in cultured cells, but it has also been shown that more than 50% of the mutations in Parkin and PINK1 linked to PD are associated with defects in mitophagy.47 Additionally, in vivo data suggests that mitophagy failure affects the health of DA neurons and plays a role in the etiology of PD. Loss of endogenous Parkin causes specific DA neurotoxicity and L-DOPA reversible motor impairment, and phosphorylation of ubiquitin at Ser65, a hallmark of PINK1 activation during mitophagy, was enhanced in a mutant animal with mitochondrial malfunction. Mitophagy activation in DA neurons was found to be age-dependent and Parkin/PINK1 dependent in studies employing a Drosophila model that expressed mitophagy probes. Depleting the deubiquitinase UPS30 in flies may restore Parkin/PINK1-dependent mitophagy and mitigate paraquat-induced motor dysfunction and reduced dopamine levels, which are symptoms similar to PD, according to another research. Other PD genes, including as α-synuclein, LRRK2, and GBA, may also contribute to PD pathogenesis through their effects on mitophagy, as will be explored later on, in addition to Parkin and PINK1, which are essential for mitophagy.47 As PINK1 activity biomarker pS65-Ub accumulates in the elderly, it may be deduced that there is an abnormality in PINK1-dependent mitophagy caused by aging. Aging PD brain tissues showed further pathological signs of altered mitophagy, including elevated levels of mitochondrial matrix protein, ATP synthase subunit β (ATP5β), and the OMM protein Miro. It is possible that mitophagy initiation is compromised when Miro is upregulated, leading to increased mitochondrial motility. Hence, the abnormal buildup of ATP5ο and Miro in the brain after death suggests that PD is linked to defective mitophagy. Platelets from people with PD showed decreased mitophagy and lower levels of LC3II and MsrB2, two factors that influence Parkin methionine oxidation, when contrasted with controls of the same age. Defective mitophagy is the likely cause of these pathological and clinical manifestations in PD.48
Parkin-independent mitophagy
Increasing evidence indicates that Parkin is not canonically necessary for all mitophagy processes. The capacity of certain protein receptors to bind to LC3 and induce mitophagy through the receptor-mediated pathway is dependent on the presence or absence of Parkin.49 This interaction is known as the LC3-interacting region (LIR) motif. In the absence of Parkin61, AMBRA1 is able to induce mitophagy via interacting with LC3 through the LIR motif. There is evidence that FUNDC1 induces hypoxia-related mitophagy through its LIR domain relationship with LC3, MARCH5 and ULK1 contacts, and FUNDC1 itself begins mitophagy independently of Parkin. One type of outer mitochondrial membrane protein that may carry out mitophagy independently of Parkin is BNIP3L, which stands for Bcl-2/adenovirus E1B 19kD-interacting protein 3. Another name for this protein is NIX. Researchers found that BNIP3L/NIX-mediated mitophagy preserved mitochondrial activity in PD patient cells, even though the cells carried non-functional Parkin.44 This finding implies that the neuroprotection is related to PD. Recently, STX1767, FKBP868, Bcl2-L-1339, and PHB269 were added to the list of developing proteins that operate as receptor-mediated mitophagy interactors. Cardiolipin is a lipid that, like the receptors stated before, externalizes from intracellular membrane to extracellular matrix (OMM) in response to mitochondrial stress and binds to LC3.50 At a certain level, this binding initiates the induction of mitophagy inside neurons. It has also been proposed that in parkin-independent, lipid-mediated mitophagy, the sphingolipid ceramide can bind autophagolysosomes to mitochondrial membranes via LC3 contacts. PINK1’s capacity to induce mitophagy through NDP52 and optineurin in the absence of Parkin allowed the identification of ubiquitin-mediated mitophagy, recasting Parkin’s function as a signal amplifier rather than an essential regulator.51 Mitophagy activation at damaged mitochondria and in a Parkin-independent route leading to mitochondrial ubiquitination and mitophagy are facilitated by interactions between mitofusin (Mfn) and other E3 ligases, such as MUL1. To facilitate PINK1-dependent mitophagy through mitochondrial ubiquitination in a way that is unrelated to Parkin, Synphilin-1 recruits SIAH1, another E3 ligase. Taken together, these findings point to a complicated mitophagy system with a multi-level compensatory network that reacts to different homeostatic and pathological states. It is possible to repair mitophagy deficits and rescue neurodegeneration in PD by using these alternative mitophagy mechanisms, such as MUL1- or NIX-involved mitophagy, even if there is minimal evidence of Parkin-independent mitophagy being implicated in PD.52 Fig. 3
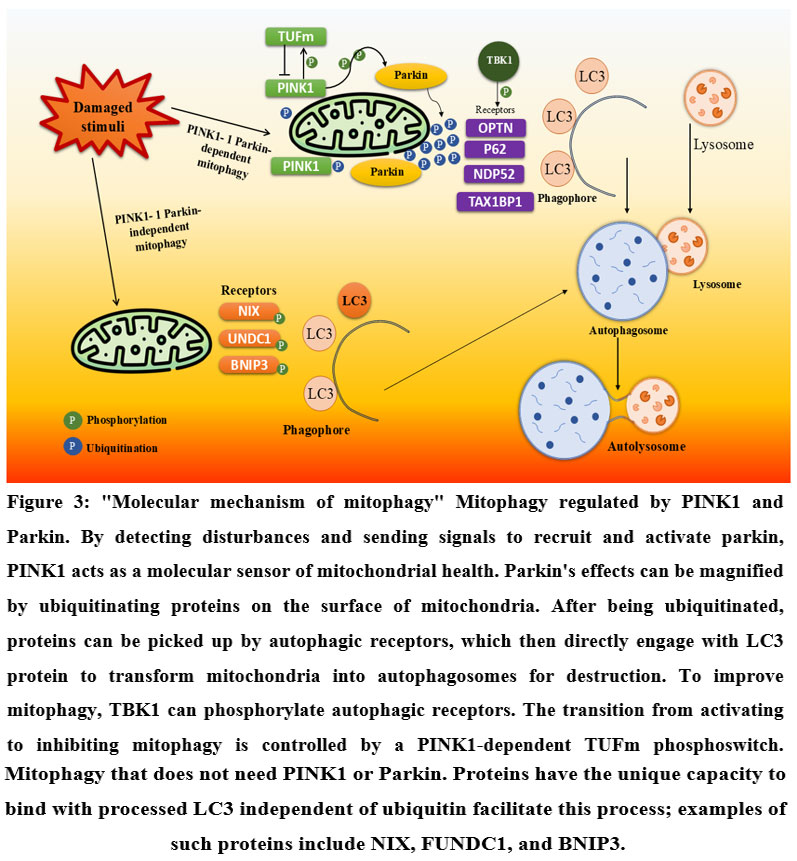 |
Figure 3: “Molecular mechanism of mitophagy” Mitophagy regulated by PINK1 and Parkin. |
(PINK1: PTEN-induced kinase 1, Parkin: Parkin RBR E3 ubiquitin-protein ligase, TUFm: Mitochondrial Tu translation elongation factor, TBK1: TANK-binding kinase 1, OPTN: Optineurin, P62: Sequestosome 1 (SQSTM1), NDP52: Nuclear dot protein 52 kDa, TAX1BP1: Tax1-binding protein 1, NIX: BNIP3-like protein X, UNDC1: Unc-51 like autophagy activating kinase 1 domain-containing 1, BNIP3: BCL2/adenovirus E1B 19 kDa protein-interacting protein 3, LC3: Microtubule-associated protein 1A/1B-light chain 3)
Mitophagy in Neurons
Mitophagy is a form of selective autophagy that facilitates the destruction of dysfunctional or surplus mitochondria. Impaired mitophagy results in mitochondrial dysfunction, which is linked to the etiology of various neurodegenerative disorders.53 Mitophagy is crucial for neurons due to their elevated energy requirements and more intricate mitochondrial structure compared to other cell types. Mitophagy eliminates small, damaged mitochondria, triggered by a distal linear wave-like phenomena extending from the soma to the axon.54 The neuronal cell body houses the majority of mitochondria, whereas the axon has a limited number of mitochondria. Consistent with mitochondrial dynamics, functioning mitochondria transit from the soma to the axon, while impaired mitochondria are retrogradely transported from the axon to the cell body. Approximately 75% of mitophagy transpires in the soma, whereas only 10–20% of mitochondrial breakdown occurs in dendrites and axon terminals. A decline in mitophagy associated with age has been observed in Drosophila, rats, and humans. Nonetheless, the critical function of mitophagy in neurons exposed to PD-related stressors requires further examination.55
The E3 ligase Parkin facilitates the ubiquitination of OLFM1 on lysosomes, and the ensuing elongation of the ubiquitin chain subsequently attracts and mediates the recruitment of p62 and LC3 to OLFM1 aggregates.56 OLFM1 attracts and concentrates deleterious mitochondria at lysosomes through indirect interaction with Dj-1 and p62, which then facilitates mitochondrial breakdown via colocalized lysosomes. OLFM1 oligomerization recruits and sensitizes TRPM2 channels to Ca2+ influx, resulting in the activation of calpain 1 and subsequent cleavage of OLFM1, which disrupts the lysosome-OLFM1-PLIN2-mitochondria link, hence facilitating mitochondrial recovery. An alternative ubiquitin-independent route enhanced by Parkin was discovered. This mechanism facilitates the recruitment and degradation of depolarized mitochondria and operates in conjunction with the ubiquitin-dependent system to augment the breakdown of damaged mitochondria by encouraging lysosomal fusion with bigger mitochondria.57 Subsequent investigations aimed at identifying compartments indicated the discovery of a new salvage route that facilitates the time- and proteasome-dependent repair of damaged lysosomal membranes. Poly-ubiquitylation of the damaged lysosomal Rift facilitates its retrotranslocation to the cytosol, enabling its degradation by the proteasome.58
Dysregulation of Mitophagy in PD
The disease processes of PD remain partially understood; nevertheless, a prevalent molecular etiology in sporadic PD is believed to entail oxidative stress-induced impairment of the ubiquitin-proteasome system (UPS) and the autophagy-lysosome-mediated macroautophagy pathways.59 Alongside its characterisation in autophagy, there has been a growing body of research examining the involvement of mTOR and its regulatory components in the pathophysiology of PD. Cellular and animal models provide consistent evidence that mTOR hyper-activation may act as a central mechanism by which diverse genetic factors associated with PD disrupt cellular proteostasis.59
In the presence of nutrients and growth factors, mTOR is activated and phosphorylates downstream targets that facilitate cell development, including S6Ks and eukaryotic initiation factor 4E-binding proteins (4E-BPs), thus enhancing mRNA translation.60,61 In response to stresses like extended nutrient scarcity, hypoxia, and oxidative stress, mTOR is inactivated by various nutrient and growth factor sensing kinases, resulting in the de-phosphorylation and activation of the downstream Forkhead box O (FoxO) transcription factors.62 These components, together with other signal regulators, actively facilitate autophagic downstream processes, including the destruction of ubiquitin-conjugated protein aggregates and Parkin-mediated mitophagy.63
Upon mitochondrial import, PINK1 detects mitochondrial depolarization and recruits Parkin from the cytosol to the depolarized mitochondria. The cascade commences with the phosphorylation of ubiquitin by mitochondrial PINK1, subsequently leading to the extensive ubiquitination of outer mitochondrial membrane proteins by Parkin. Ubiquitinated outer mitochondrial membrane proteins are identified by ubiquitin adapter proteins, such as p62, which is crucial for mitochondrial incorporation into phagophores.64 The closing of phagophores is a crucial phase in their growth into fully developed autophagosomes. Subsequent to closure, the resultant autophagosomes, remaining within the cytosol, experience changes to cytosolic proteins, including phosphorylation and ubiquitylation. The two steps lead to the recruitment of the cytosolic SEC62 subcomplex and its upstream scaffold proteins, which are crucial for lysosomal fusion, lysosomal trafficking, and amphisome formation.65
Crosstalk Between mTOR and Mitophagy
Mitochondrial dysfunction is increasingly associated with the etiology of neurodegenerative illnesses, such as PD, wherein the resultant loss of striatal dopaminergic neurons leads to various neuropsychiatric and motor impairments. Comprehending the principles of mitophagy, which selectively eliminates damaged mitochondria, is crucial for devising innovative therapeutic techniques to address neurological disorders linked to mitochondrial malfunction.66 Evidence indicates that the interaction between the mechanistic target of rapamycin signalling pathway and mitophagy may be a double-edged sword in PD. The anomalous activation of the mechanistic target of rapamycin signalling pathway causes hyperphosphorylation of the serine/threonine protease homeodomain interacting protein kinase-2 and suppresses PINK1 protein production, resulting in the impairment of the PINK1/parkin signalling pathway. Inhibition of the mechanistic target of rapamycin pathway through genetic modification or pharmacological activation mitigates neurodegeneration of dopaminergic neurons in PINK1 or parkin mutants and enhances motor performance.15 Decreased neuroinflammation in the brain is noted in organotypic cortical slices treated with pharmacological drugs, linked to the inhibition of mTORC1 and upstream p62/SQSTM1.67 The impact of the mechanistic target of rapamycin pathway on the etiology of PD is contingent upon the experimental paradigms employed. The G2019S mutation of the leucine-rich repeat kinase 2 is the predominant cause of familial PD. LRRK2 governs many signaling pathways in reaction to environmental stimuli and modulates cellular processes essential for neuronal integrity. LRRK2 is proposed to be recruited to damaged mitochondria in a parkin-dependent fashion, and the inhibition of LRRK2 is adequate to promote mitophagy and alleviate neurodegeneration in parkin-deficient animals. Following mitochondrial impairment, mTORC1 phosphorylates serine 196 of Drp1 on the injured mitochondria, facilitating its translocation to the mitochondria and enhancing mitophagy. Conversely, mTORC2 phosphorylates Drp1 at serine 636, inhibiting DRP1 GTPase activity and the initiation of mitophagy. Inhibiting D2R-mediated mTORC1 activation induces mitophagy, increases mitochondrial mass, and enhances motor capabilities in mice overexpressing α-synuclein, whereas mTORC1 activation alone can replicate PD-like symptoms.68
mTOR’s Influence on Mitophagy
Mitochondrial dynamics, encompassing membrane fission and fusion, mitophagy, and transport, regulate bioenergetic balance and cellular death by modulating mitochondrial morphology and localization. Recent research has highlighted the relationship between mitochondrial dynamics and neurodegenerative disorders, indicating that mitochondrial damage and abnormal dynamics precede organelle fragmentation and depolarization loss, suggesting that early perturbations in mitochondrial dynamics are critical events in neurodegeneration.69 Comprehending how signaling networks govern mitochondrial dynamics may yield significant insights into the pathogenesis of neurodegenerative diseases. Multiple lines of evidence suggest that abnormal mitochondrial fission and mitophagy contribute to dopaminergic neuronal damage in PD. α-Synuclein facilitates mitochondrial fission and impedes fusion, resulting in oxidative stress-induced mitochondrial fragmentation. The activation of the mTOR pathway may impede mitochondrial autophagy and intensify damage mediated by mitochondrial fission in PD. Moreover, many treatment targets that augment these pathways may safeguard midbrain dopaminergic neurons against mitochondrial stress in experimental models of PD.70
Mitophagy is the mechanism via which impaired or nonfunctional mitochondria are designated for destruction by the lysosomal system. Mitophagy encompasses the start, selection, and degradation phases. During the initiation phase, the mitochondrion is designated as cargo for engulfment by the phagophore. This procedure necessitates the demembranation of the OMM, leading to the liberation of crucial mitophagy signalling molecules and the exposure of ubiquitination substrates on the OMM.71 Puma and Ninjurin1 (NINJ1) are among the initial proteins recognized for their role in inducing mitophagy through the demembranation of the OMM. Mitochondrion-associated ubiquitin E3 ligases (Parkin and RING-finger 1), defects in mitochondrial respiratory chain complex I, and demembranation triggered by BAX/BAK-dependent mitochondrial outer membrane permeabilization (MOMP) or ceramide accumulation contribute to the activation of supplementary pathways for the early selection of depolarized mitochondria. These initiators can either directly mark mitochondria by ubiquitin modification or indirectly facilitate the ubiquitination cascade by degrading mitochondria-associated scaffold proteins. During the initial selection phase, outer mitochondrial compartment proteins (Miro1, Mfn1, Mfn2, OPA1, Parkin, or VDAC) are either attracted to the dysfunctional mitochondrion or modified in their conformation by interaction with upstream mitophagy signalling molecules. The selection process is crucial in determining the destiny of mitochondria.72
Mitophagy’s Impact on mTOR Activity
Mitophagy, a selective form of autophagy, is regulated by increased glycolysis, resulting in the recruitment of damaged mitochondria. Significantly, mitophagy can stimulate mTORC1 activity in both yeast and human cells. Conversely, the silencing and inhibition of mitophagy-related genes obstruct the decrease of mTORC1 activity induced by therapy. The findings indicate that mitophagy can modulate mitochondrial dynamics and mTOR activity, potentially safeguarding against the advancement of age-related neurological disorders.73
Mitophagy may enhance mitochondrial dynamics and mTOR activity levels, hence providing protection. Mitophagy selection activates the binding domain that recruits the E3 ligase to impaired mitochondria, facilitating the ubiquitination of proteins.74 Ubiquitinated mitochondrial proteins can adjust to mitophagy substrates. The most prevalent substrate protein in the outer mitochondrial membrane is recruited and destroyed through mitophagy. Attaches to mitochondria to impede mitophagy; however, ubiquitination secures it in an autophagic state, which ceases binding. As a result, mitophagy transpires and mitochondrial morphology is modified.71
A lag phase in mitophagy signifies a possible target for therapeutic intervention. may temporarily inhibit. Prolonged suppression results in mitochondrial fragmentation and the release of pro-apoptotic proteins, ultimately causing neuronal death.73 Excessive mitophagy, like its severe inhibition, is deleterious, as evidenced by mitochondrial morphologies and viability. Auto-mitophagy can induce significant disruption, and a reduction of intracellular proximal mitochondria below a critical concentration triggers mTOR activation. Mitophagy may not negate the therapeutic potential of mTOR inhibition.12
Bidirectional Regulation
Mitophagy, the selective destruction of mitochondria in reaction to changes in metabolism, is a prominent example of physiological crosstalk between two processes. By suppressing PGC1α and ALD, the mTOR signaling pathway negatively controls mitochondrial biogenesis.75 On the flip side, mTORC1 and mTORC2 drive mitochondrial breakdown and dysfunction through favorably regulating Parkin-mediated mitophagy through boosting its translocation and activation. That mTOR signaling can promote mitophagy or increase mitochondrial biogenesis depends on the context is suggested by this.76 Inhibition of mTORC1 activity occurs when low-energy signals activate AMPK, leading to an increase in TSC2 phosphorylation. The enhancement of PGC1α activity and protein stability leads to mitochondrial biogenesis. On the other hand, AMPK can catalyze the elimination of damaged mitochondria by directly phosphorylating ULK1, a key activator of the conventional mitophagy pathway. Parkin and PINK1 would work together to ubiquitinate the OMM protein VDAC1 at a crucial spot close to the bilayer curvature in reaction to this conflict.76 In order to reveal the locations where Parkin targets, this would enhance mitochondrial fission, fragmentation, and unfolding. Parkin is phosphorylated at Ser108 by ULK1 during translocation. This improves Parkin’s ubiquitination and degradation-inhibiting capabilities toward VDAC1.77 Furthermore, mitochondrial dysfunction is often accompanied by selective degradation of defective processes and biogenesis abnormalities; mutations in PINK1 or Parkin would lead to an aberrant buildup of enlarged mitochondria, a hallmark of sporadic PD.78 Importantly, the amount of mitochondrial malfunction and the concentration of mitochondrial toxic P-tau appear to correlate with the survival or death of neurons. Significant amounts of damage cause the slow loss of important regulators like PGC1α, TFAM, and AMPK, which leads to various harmful aggregates or at least maintains a low ratio of these processes. To the contrary, degrading mechanisms have very little control over mitochondrial biogenesis; thus, increasing its expression would postpone (and decrease it would reverse) the buildup of harmful P-tau aggregates. One of the key mechanisms of cognitive deterioration in AD is global energy depletion, which can be caused by excessive selectivity and degradation. In addition, there is neuron-specific preference for two critical phosphorylatin sites: S108 in Parkin and S62 in PINK1. These sites directly influence the input of phosphorylation signals. This illustrates the correlation between the presence of mitobiogenic and mitophagic processes in control astrocytes and the observed rise in mitochondrial breakdown in neurons with α-synuclein mutation in familial PD. To summarize, in order to fully comprehend the intricate link between upstream signaling networks and selective processes, additional groundbreaking findings are necessary.79
Therapeutic Targets
The dynamic characteristics of post-translational modifications, lipid/protein membranes, and endo-lysosomal compartments linked to mitochondria must be thoroughly examined to accurately define and address the complexities of this dual-faceted phenomenon in mitophagy/mTOR signaling pathways, along with interconnected pathways in PD and related neuropathologies 80. The ongoing discovery of therapeutics aimed at mTOR signaling to regulate cell death and survival is currently being pursued for AD and other neurodegenerative disorders. The increasing utilization of CRISPR/Cas9 gene editing technology is anticipated to facilitate temporal modulation of the expression of targeted pathogenic proteins. Proactive investigations into interventions targeting the specified mitophagy-regulatory molecules/proteins, whether equal to or positioned upstream or downstream of Pink1/Parkin, should yield additional pathways for therapeutic advancement in PD, tauopathies, and mitochondrial metabolic syndrome/metabolic dysfunction. Although mTOR signalling and mitophagy represent emerging fields of investigation in PD, associated neurodegenerative disorders, and injuries, additional research on the intercommunication of multi-directional pathways among these processes and others should enhance our comprehension of disease pathogenesis and mechanisms, while also promoting the potential application of mTOR signalling and mitophagy as targeted therapeutic strategies. Comprehensive investigations of mTOR signalling and mitophagy in various mammalian models or human cell-based models of PD/pathogenesis will significantly enhance our understanding of the roles of these processes in cell death and survival under diverse chemical stresses, as well as the relative precedence of each process in facilitating contrasting cellular functions 81. It is becoming increasingly evident that, with the sort of “insult,” targeted the relative priorities and dominance of mitophagy and mTOR signalling regulate cellular activities. Augmenting early-onset mTOR activity along the gray/red axis may serve as a targeted cell-revival strategy to prevent or reverse age-related neurodegeneration. Consequently, comprehensive advancement in targeting the mTOR/mTOR-Akt/mitophagy pathways in nursing contexts unrelated to PD, neurodegeneration, or accidents necessitates the resolution of these and other significant concerns.82
Table 1: Therapeutic drugs that affect mitophagy and mTOR signaling pathways in PD
| Group | Treatment | Mechanism of Action | Effect on mTOR | Effect on Mitophagy | Reference |
| AMPK Activators (Indirect mTOR Inhibitors) | Metformin | Activates AMPK → inhibits mTOR | Inhibits via AMPK | Promotes | 83 |
| Resveratrol | Activates SIRT1 and AMPK → indirect mTOR inhibition | Indirect inhibition | Enhances | 84 | |
| Berberine | Activates AMPK → inhibits mTOR | Inhibits via AMPK | Promotes | 83 | |
| Quercetin | Activates AMPK + direct mTOR inhibition | Inhibits | Enhances | 85 | |
| SIRT1 Activators / NAD+ Boosters | Resveratrol | Activates SIRT1 and AMPK | Indirect inhibition | Enhances | 84 |
| Nicotinamide Riboside | NAD+ precursor → activates SIRT1 | Indirect modulation | Promotes | 86 | |
| Direct mTOR Inhibitors / Modulators | Rapamycin | Direct mTORC1 inhibition | Inhibits mTORC1 | Enhances | 87 |
| Curcumin | Direct mTOR inhibition | Inhibits | Enhances | 87 | |
| EGCG | Downregulates mTOR | Downregulates | Promotes | 88 | |
| Baicalein | Suppresses mTOR signaling | Suppresses | Promotes | 88 | |
| Ginsenosides (Rg1, Rb1) | Modulate mTOR & autophagy | Modulate | Induce | 88 | |
| mTOR-Independent Autophagy/Mitophagy Inducers | Trehalose | Autophagy induces via mTOR-independent mechanisms | No direct effect | Promotes | 89 |
| Urolithin A | Activates PINK1/Parkin pathway → direct mitophagy induction | No direct modulation | Induces | 89 | |
| Sulforaphane | Activates Nrf2 pathway → modulates autophagy | May inhibit indirectly | Enhances | 90 | |
| Mitochondrial Electron Transport / Supporters | Coenzyme Q10 | Supports mitochondrial electron transport | No direct effect | Supports indirectly | 91 |
| GSK-3β Inhibitors / Other Pathways | Lithium | Inhibits GSK-3β → modulates autophagy | Partial inhibition | Enhances | 92 |
Challenges in Treatment
The dual nature of nutrient-sensing pathways and oxidative phosphorylation mechanisms determines their intricate function in pathogenicity, thus offering a chance for experimental investigation to selectively direct these mechanisms towards therapeutic applications. The impact of manipulating either pathway on mitophagy outcomes in both in vitro and in vivo models pertinent to PD must be examined. This is particularly useful for the prospective application of bipolar mTOR regulation as a therapeutic intervention to prevent and/or mitigate PD-like symptoms in animal models, as well as for usage in cellular models during experimental validation.93
Knowledge accumulated regarding mTOR-related pathways must be used to clinical research including human cohorts. Implementing a significant study design is difficult due to the complex and often interdependent structure of these pathways, indicating that modifications in one route may be obscured by changes occurring upstream. At a minimum, specific phosphoproteomic methods are necessary to assess the direct activation or repression of particular pathway components. Nonetheless, isolating these individual pathway components is improbable to yield successful identification of PD-specific consequences, especially concerning mTORC2/PP2A signalling. Consequently, a thorough examination of altered circadian rhythms and their effects on intermediate metabolic control may assist in addressing this difficulty.7,94
Expanding the neurodegenerative illness cohort to encompass more tauopathies, LINCL, or other lysosomal dysfunctions may reveal common mitochondrial signalling interactions that are not exclusive to the etiology of tau or PD pathology. Furthermore, exploring other mechanisms beyond TSC1/TSC2 or GSK-3β regulation of mTORC1 may be advantageous. Finally, existing information gaps regarding the influence of food signals on the precision of the circadian clock system remain unexplored and may offer potential for retargeting initiatives.95
Future Directions
Despite extensive study by academia and business over several decades, no disease-modifying medication for PD has effectively entered clinical use to yet. One strategy being implemented is the creation of therapies designed to restore or improve mitochondrial quality control, utilizing either small-molecule pharmaceuticals or gene therapy. This line of inquiry has demonstrated a connection between genetic abnormalities affecting the E3 ligase Parkin and the onset of autosomal recessive PD. Considering that Parkin activity necessitates the upstream recruitment of the kinase PINK1, considerable work has been dedicated to developing modulators of the PINK1–Parkin pathway as disease-modifying therapies for PD. The recent advancements in the in vivo delivery of small compounds and AAV-mediated control of PINK1 and Parkin highlight the promise of this methodology. Nonetheless, akin to most accumulation disorders, there are obstacles in turning this approach into a clinically applicable treatment for PD, many of which pertain to target selection and the predictive capacity of model systems.96
Examining how neurons with effective autophagic mechanisms may endure dysfunctional mitochondria may yield pharmacological probes and genetic tools to delineate and ultimately enhance mitophagic pathways pertinent to human disease.12 Identified compounds should be evaluated for selectivity in human cells, as selective mitophagy may be modulated by small molecules. These initiatives align with the continuous identification of supplementary signaling molecules and pathways pertinent to PINK1–Parkin recruitment, an enhanced understanding of mitochondrial receptors implicated in the recruitment of alternative mitophagic pathways, and the integration of PINK1–Parkin mitophagy with macroautophagy.10 These pathways represent under-explored and clinically actionable opportunities for therapeutic intervention in PD. Furthermore, when a therapeutic target is identified, target validation studies employing chemical probes or gene therapy methodologies can effectively delineate the correlation between target engagement and biological outcomes in mid- to high-throughput settings. Irrespective of the original methodology employed, a definitive output of existing knowledge is essential, and comprehensive pathway maps illustrating the current interactions within the principal mitophagy pathways will facilitate further advancement. Simultaneously, it is imperative to guarantee that the aforementioned concepts are implemented in the exploration of other possible therapeutic targets.97
Innovative Therapeutic Strategies
The rising life expectancy in developed nations results in an escalating burden of age-related neurological disorders, particularly PD. PD is characterized by the gradual accumulation of the α-syn protein into insoluble aggregates known as Lewy bodies. The aggregation of α-syn is facilitated by several genetic factors, environmental pollutants, metabolic irregularities, and neuroinflammatory processes associated with age, leading to a complicated etiology that remains inadequately comprehended.98 It is predicted that around four million individuals globally are afflicted with PD, a figure projected to quadruple by 2030. Although several pathophysiological mechanisms characterize this group of illnesses, a unifying element is the impairment of one or more fundamental cellular pathways related to proteostasis, resulting in protein misfolding, aggregation, and toxicity.
PD is a multifaceted and heterogeneous neurodegenerative disorder associated with SNCA, impacting both the central and peripheral nervous systems, resulting in motor and non-motor symptoms.99 The clinical manifestations of PD differ in terms of progression rate and certain motor and non-motor symptoms, which remain to be elucidated. The pathogenesis of PD is primarily characterized by the aggregation of insoluble fibrillar α-synuclein aggregates forming Lewy bodies in a statistically skewed distribution throughout various brain regions over time. Increasing data suggests that this process can be initiated and disseminated in a prion-like fashion, highlighting the potential influence of local or systemic factors in the onset and/or propagation of PD. The α-synuclein cells release and absorb potential extracellular vesicles (EVs), leading to abnormal α-synuclein proliferation both in vitro and in vivo. Lewy bodies comprise fibrillar α-synuclein protein and are associated with various structural and biochemical alterations in the cytoplasm. To mitigate α-synuclein aggregation, enhance clearance, and restore proteostasis, cells depend on numerous compensatory mechanisms, many of which remain inadequately described. The mechanisms of α-synuclein clearance examined include molecular chaperones, proteasomes, the autophagy-lysosome pathway, and extracellular vesicles.100,101
Conclusion
Increasing evidence indicates that dysregulation of the mechanistic/mammalian target of rapamycin is linked to neurodegeneration, particularly in neurodegenerative disorders. In recent years, the involvement of mTOR in the pathophysiology of PD has been extensively examined. mTOR is a conserved serine/threonine kinase that governs various cellular processes essential for metabolism, proliferation, growth, and survival. mTOR signaling is modulated by various endogenous and external variables, including growth factors, cellular energy levels, nutrition, and mitochondrial function. It is categorized into two separate multi-protein complexes that vary in their subunit makeup and subsequent signaling pathways. An increasing amount of evidence indicates that mTORC1 dysfunction in neurons plays a role in the etiology of PD. In PD, mTORC1 hyperactivity is predominantly associated with compromised autophagy, either through direct interactions with autophagy-related proteins or indirectly via pathways involving oxidative stress, inflammation, and neurotrophic factors. Hyperactivity of mTORC1 thus facilitates the development of Lewy bodies and the degeneration of dopaminergic neurons. In contrast, mTORC1 activity is diminished in the substantia nigra of PD mice models and in low tau variant iPSC-derived neurons. Inhibition of mTORC1 impedes the elimination of pathogenic tau. mTORC2 is also recognized for its function in PD. Enhanced mTORC2 activity in SH-SY5Y cells is noted after exposure to the parkinsonian toxin, whereas mTORC2 inhibition exacerbates the toxic consequences. Both acute and chronic exposure decreases mTORC2 signaling in vivo.
Through the transcription factor TFEB, mTORC1 not only controls translation but also plays an important negative regulatory role in lysosome formation and activity. So, blocking mTORC1 improves neuronal lysosomal biogenesis and function, which includes heightened activity in chaperone-mediated and macroautophagic pathways, reduced α-synuclein aggregation, and removal of harmful protein aggregates. On the other hand, ±-synuclein degradation is reduced, aggregation and toxicity are increased, and lysosomal function is impaired due to mTORC1 hyperactivity. Additional investigation into the involvement of mitochondria in controlling mTORC1 activity in PD models is warranted, among other endogenous and external variables. The control of mTORC1 after mitochondrial depolarization is associated with PINK1 and Parkin. In inherited types of PD, neurodegeneration and impaired selective mitochondrial clearance are caused by defects in the function of either PINK1 or Parkin.
Acknowledgement
Authors are thankful to Chandigarh University (University Institute of Pharma Sciences) for providing excellent support to complete this work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials
Permission to reproduce material from other sources
Not Applicable
Author Contributions
- Ritika Sharma: Writing original draft and reference
- Hitesh Dewangan: Conceptualization, supervision, critically review and editing
- Kundan Bora– Conceptualization, supervision, critically review and editing
Reference
- Khan AU, Akram M, Daniyal M, Zainab R. Awareness and current knowledge of PD: a neurodegenerative disorder. International Journal of Neuroscience. 2019;129(1):55-93.
CrossRef - Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nature Reviews Neurology. 2016;12(7):379-392.
CrossRef - Lan A-p, Chen J, Zhao Y, Chai Z, Hu Y. mTOR signaling in PD. Neuromolecular medicine. 2017;19:1-10.
CrossRef - Subramanian I, Pushparatnam K, McDaniels B, Mathur S, Post B, Schrag A. Delivering the diagnosis of PD-setting the stage with hope and compassion. Parkinsonism & Related Disorders. 2024;118
CrossRef - Jankovic J, Lang AE. Diagnosis and assessment of PD and other movement disorders. Bradley’s Neurology in Clinical Practice E-Book. 2021;310(1).
- Moore MN. Lysosomes, autophagy, and hormesis in cell physiology, pathology, and age-related disease. Dose-Response. 2020;18(3):1559325820934227.
CrossRef - Sharma R, Kour A, Dewangan HK. Enhancements in PD Management: Leveraging Levodopa Optimization and Surgical Breakthroughs. Current Drug Targets. 2025;26(1):17-32.
CrossRef - Sinnige T, Stroobants K, Dobson CM, Vendruscolo M. Biophysical studies of protein misfolding and aggregation in in vivo models of Alzheimer’s and PDs. Quarterly Reviews of Biophysics. 2020;53:e10.
CrossRef - Smith LJ, Lee C-Y, Menozzi E, Schapira AH. Genetic variations in GBA1 and LRRK2 genes: Biochemical and clinical consequences in PD. Frontiers in Neurology. 2022;13:971252.
CrossRef - Barazzuol L, Giamogante F, Brini M, Calì T. PINK1/parkin mediated mitophagy, Ca2+ signalling, and ER–mitochondria contacts in PD. International journal of molecular sciences. 2020;21(5):1772.
CrossRef - Masaldan S, Callegari S, Dewson G. Therapeutic targeting of mitophagy in PD. Biochemical Society Transactions. 2022;50(2):783-797.
CrossRef - Lu Y, Li Z, Zhang S, Zhang T, Liu Y, Zhang L. Cellular mitophagy: mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13(2):736.
CrossRef - Raza A, Raina J, Sahu SK, Wadhwa P. Genetic mutations in kinases: a comprehensive review on marketed inhibitors and unexplored targets in PD. Neurological Sciences. 2025:1-16.
CrossRef - Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Current Biology. 2018;28(4):R170-R185.
CrossRef - Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nature Reviews Neuroscience. 2011;12(8):437-452.
CrossRef - Pang SY-Y, Ho PW-L, Liu H-F, et al. The interplay of aging, genetics and environmental factors in the pathogenesis of PD. Translational Neurodegeneration. 2019;8:1-11.
CrossRef - Chu CT. A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for PD. Human molecular genetics. 2010;19(R1):R28-R37.
CrossRef - Bej E, Cesare P, Volpe AR, d’Angelo M, Castelli V. Oxidative stress and neurodegeneration: insights and therapeutic strategies for PD. Neurology International. 2024;16(3):502-517.
CrossRef - Tafur L, Kefauver J, Loewith R. Structural insights into TOR signaling. Genes. 2020;11(8):885.
CrossRef - Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Science signaling. 2009;2(84):pe51-pe51.
CrossRef - Deleyto-Seldas N, Efeyan A. The mTOR–autophagy axis and the control of metabolism. Frontiers in cell and developmental biology. 2021;9:655731.
CrossRef - Hu X, Guo F. Amino acid sensing in metabolic homeostasis and health. Endocrine reviews. 2021;42(1):56-76.
CrossRef - Chrienova Z, Nepovimova E, Kuca K. The role of mTOR in age-related diseases. Journal of Enzyme Inhibition and Medicinal Chemistry. 2021;36(1):1678-1692.
CrossRef - Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nature reviews Molecular cell biology. 2020;21(4):183-203.
CrossRef - Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nature reviews immunology. 2012;12(5):325-338.
CrossRef - Alers S, Löffler AS, Paasch F, et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy. 2011;7(12):1424-1433.
CrossRef - Wong P-M, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9(2):124-137.
CrossRef - Zaki ES, Sayed RH, Saad MA, El-Yamany MF. Roflumilast ameliorates ovariectomy-induced depressive-like behavior in rats via activation of AMPK/mTOR/ULK1-dependent autophagy pathway. Life sciences. 2023;327:121806.
CrossRef - Wang Y, Zhang H. Regulation of autophagy by mTOR signaling pathway. Autophagy: Biology and diseases: Basic science. 2019:67-83.
CrossRef - Xu G, Zhang Q, Cheng R, Qu J, Li W. Survival strategies of cancer cells: the role of macropinocytosis in nutrient acquisition, metabolic reprogramming, and therapeutic targeting. Autophagy. 2025;(just-accepted)
CrossRef - Salminen A, Kaarniranta K, Kauppinen A, et al. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Progress in neurobiology. 2013;106:33-54.
CrossRef - Querfurth H, Lee H-K. Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Molecular neurodegeneration. 2021;16(1):44.
CrossRef - Błaszczyk JW. Energy metabolism decline in the aging brain—pathogenesis of neurodegenerative disorders. Metabolites. 2020;10(11):450.
CrossRef - Li W, He P, Huang Y, et al. Selective autophagy of intracellular organelles: recent research advances. Theranostics. 2021;11(1):222.
CrossRef - Zhang K, Li Q, Zhang Y, et al. Targeting Mitophagy as a Potential Therapeutic Approach for Age‐Related Bone Diseases. Advanced Therapeutics. 2024;7(7):2400078.
CrossRef - Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antioxidants & redox signaling. 2012;17(5):794-802.
CrossRef - Palmieri F. Diseases caused by defects of mitochondrial carriers: a review. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2008;1777(7-8):564-578.
CrossRef - Montero J, Mari M, Colell A, et al. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2010;1797(6-7):1217-1224.
CrossRef - Mejia EM, Hatch GM. Mitochondrial phospholipids: role in mitochondrial function. Journal of bioenergetics and biomembranes. 2016;48:99-112.
CrossRef - Li S, Lei Z, Sun T. The role of microRNAs in neurodegenerative diseases: a review. Cell biology and toxicology. 2023;39(1):53-83.
CrossRef - Yadav E, Yadav P, Khan MMU, Singh H, Verma A. Resveratrol: A potential therapeutic natural polyphenol for neurodegenerative diseases associated with mitochondrial dysfunction. Frontiers in Pharmacology. 2022;13:922232.
CrossRef - Reddy DS, Abeygunaratne HN. Experimental and clinical biomarkers for progressive evaluation of neuropathology and therapeutic interventions for acute and chronic neurological disorders. International Journal of Molecular Sciences. 2022;23(19):11734.
CrossRef - Eldeeb MA, Esmaili M, Hassan M, Ragheb MA. The role of PTEN-L in modulating PINK1-Parkin-mediated mitophagy. Neurotoxicity Research. 2022;40(4):1103-1114.
CrossRef - Xiao B, Kuruvilla J, Tan E-K. Mitophagy and reactive oxygen species interplay in PD. npj PD. 2022;8(1):135.
CrossRef - Celtikci B. A crosstalk between dual-specific phosphatases and dual-specific protein kinases can be a potential therapeutic target for anti-cancer therapy. Protein Kinase-mediated Decisions Between Life and Death. Springer; 2021:357-382.
CrossRef - Grenier K, McLelland G-L, Fon EA. Parkin-and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Frontiers in neurology. 2013;4:100.
CrossRef - Malpartida AB, Williamson M, Narendra DP, Wade-Martins R, Ryan BJ. Mitochondrial dysfunction and mitophagy in PD: from mechanism to therapy. Trends in biochemical sciences. 2021;46(4):329-343.
CrossRef - Lee SH, Lee S, Du J, et al. Mitochondrial MsrB2 serves as a switch and transducer for mitophagy. EMBO molecular medicine. 2019;11(8):e10409.
CrossRef - Bernardini J, Lazarou M, Dewson G. Parkin and mitophagy in cancer. Oncogene. 2017;36(10):1315-1327.
CrossRef - Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Frontiers in cell and developmental biology. 2017;5:90.
CrossRef - Durcan TM, Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes & development. 2015;29(10):989-999.
CrossRef - Goiran T, Eldeeb MA, Zorca CE, Fon EA. Hallmarks and molecular tools for the study of mitophagy in PD. Cells. 2022;11(13):2097.
CrossRef - Chu CT. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiology of disease. 2019;122:23-34.
CrossRef - Surana S, Villarroel‐Campos D, Lazo OM, et al. The evolution of the axonal transport toolkit. Traffic. 2020;21(1):13-33.
CrossRef - Markaki M, Tsagkari D, Tavernarakis N. Mitophagy mechanisms in neuronal physiology and pathology during ageing. Biophysical Reviews. 2021;13(6):955-965.
CrossRef - Vainshtein A, Grumati P. Selective autophagy by close encounters of the ubiquitin kind. Cells. 2020;9(11):2349.
CrossRef - Uoselis L, Nguyen TN, Lazarou M. Mitochondrial degradation: mitophagy and beyond. Molecular cell. 2023;83(19):3404-3420.
CrossRef - Ghosh R, Vinod V, Symons JD, Boudina S. Protein and mitochondria quality control mechanisms and cardiac aging. Cells. 2020;9(4):933.
CrossRef - Limanaqi F, Biagioni F, Gambardella S, Familiari P, Frati A, Fornai F. Promiscuous roles of autophagy and proteasome in neurodegenerative proteinopathies. International journal of molecular sciences. 2020;21(8):3028.
CrossRef - Dowling RJ, Topisirovic I, Alain T, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328(5982):1172-1176.
CrossRef - Bhati V, Prasad S, Kabra A. RNA-based therapies for neurodegenerative disease: Targeting molecular mechanisms for disease modification. Molecular and Cellular Neuroscience. 2025:104010.
CrossRef - Thomas MP. Acute simulated hypoxia and ischemia in cultured C2C12 myotubes: decreased phosphatidylinositol 3-kinase (PI3K)/Akt activity and its consequences for cell survival. Stellenbosch: Stellenbosch University; 2008.
- Tsefou E, Ketteler R. Targeting deubiquitinating enzymes (DUBs) that regulate mitophagy via direct or indirect interaction with Parkin. International Journal of Molecular Sciences. 2022;23(20):12105.
CrossRef - Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Current opinion in cell biology. 2011;23(4):476-482.
CrossRef - Reggiori F, Molinari M. ER-phagy: mechanisms, regulation, and diseases connected to the lysosomal clearance of the endoplasmic reticulum. Physiological reviews. 2022;
CrossRef - Kaur S, Sharma N, Kumar V, et al. The role of mitophagy in various neurological diseases as a therapeutic approach. Cellular and Molecular Neurobiology. 2023;43(5):1849-1865.
CrossRef - Cho KS, Lee JH, Cho J, Cha G-H, Song GJ. Autophagy modulators and neuroinflammation. Current Medicinal Chemistry. 2020;27(6):955-982.
CrossRef - Limanaqi F, Biagioni F, Busceti CL, et al. Phytochemicals bridging autophagy induction and alpha-synuclein degradation in parkinsonism. International journal of molecular sciences. 2019;20(13):3274.
CrossRef - Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants. 2017;6(2):25.
CrossRef - Arenas E, Saltó C, Villaescusa C. WNT signaling in midbrain dopaminergic neuron development and cell replacement therapies for PD. SpringerPlus. 2015;4(Suppl 1):L49.
CrossRef - Xian H, Liou Y-C. Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death & Differentiation. 2021;28(3):827-842.
CrossRef - Alghamdi A. A detailed review of pharmacology of MFN1 (mitofusion-1)-mediated mitochondrial dynamics: Implications for cellular health and diseases. Saudi Pharmaceutical Journal. 2024:102012.
CrossRef - Mishra E, Thakur MK. Mitophagy: A promising therapeutic target for neuroprotection during ageing and age‐related diseases. British Journal of Pharmacology. 2023;180(12):1542-1561.
CrossRef - Peng J, Ren K-D, Yang J, Luo X-J. Mitochondrial E3 ubiquitin ligase 1: A key enzyme in regulation of mitochondrial dynamics and functions. Mitochondrion. 2016;28:49-53.
CrossRef - Zhao X, Xue X, Wang C, Wang J, Peng C, Li Y. Emerging roles of Sirtuins in alleviating alcoholic liver Disease: A comprehensive review. International Immunopharmacology. 2022;108:108712.
CrossRef - Iorio R, Celenza G, Petricca S. Mitophagy: molecular mechanisms, new concepts on parkin activation and the emerging role of AMPK/ULK1 axis. Cells. 2021;11(1):30.
CrossRef - Bayne AN, Trempe J-F. Mechanisms of PINK1, ubiquitin and Parkin interactions in mitochondrial quality control and beyond. Cellular and Molecular Life Sciences. 2019;76:4589-4611.
CrossRef - Moon HE, Paek SH. Mitochondrial dysfunction in PD. Experimental neurobiology. 2015;24(2):103.
CrossRef - Yi S, Wang L, Wang H, Ho MS, Zhang S. Pathogenesis of α-synuclein in PD: from a neuron-glia crosstalk perspective. International journal of molecular sciences. 2022;23(23):14753.
CrossRef - Mäger I, Willms E, Bonner S, Hill AF, Wood MJ. Extracellular vesicles in neurodegenerative disorders. Exosomes. Elsevier; 2020:285-305.
CrossRef - McLelland G-L. PD genes orchestrate mechanisms of mitochondrial quality control. McGill University (Canada); 2017.
- Kou X, Wu S, Kirberger M, Chen N. Exercise-Induced Autophagy and PD. Exercise, Autophagy and Chronic Diseases. 2021:155-175.
CrossRef - Steinberg GR, Carling D. AMP-activated protein kinase: the current landscape for drug development. Nature Reviews Drug Discovery. 2019/07/01 2019;18(7):527-551. doi:10.1038/s41573-019-0019-2
CrossRef - Ungurianu A, Zanfirescu A, Margină D. Sirtuins, resveratrol and the intertwining cellular pathways connecting them. Ageing Research Reviews. 2023;88:101936.
CrossRef - Chen Y-F, Qiu Q, Wang L, et al. Quercetin ameliorates myocardial injury in diabetic rats by regulating autophagy and apoptosis through AMPK/mTOR signaling pathway. The American Journal of Chinese Medicine. 2024;52(03):841-864.
CrossRef - Kang H, Park Y-K, Lee J-Y. Nicotinamide riboside, an NAD+ precursor, attenuates inflammation and oxidative stress by activating sirtuin 1 in alcohol-stimulated macrophages. Laboratory Investigation. 2021;101(9):1225-1237.
CrossRef - Kuo C-J, Huang C-C, Chou S-Y, et al. Potential therapeutic effect of curcumin, a natural mTOR inhibitor, in tuberous sclerosis complex. Phytomedicine. 2019;54:132-139.
CrossRef - Wang S, Yuan R, Liu M, et al. Targeting autophagy in atherosclerosis: advances and therapeutic potential of natural bioactive compounds from herbal medicines and natural products. Biomedicine & Pharmacotherapy. 2022;155:113712.
CrossRef - Pavlova JA, Guseva EA, Dontsova OA, Sergiev PV. Natural activators of autophagy. Biochemistry (Moscow). 2024;89(1):1-26.
CrossRef - Lu Z, Ren Y, Yang L, et al. Inhibiting autophagy enhances sulforaphane-induced apoptosis via targeting NRF2 in esophageal squamous cell carcinoma. Acta Pharmaceutica Sinica B. 2021;11(5):1246-1260.
CrossRef - Jiang YJ, Jin J, Nan QY, et al. Coenzyme Q10 attenuates renal fibrosis by inhibiting RIP1-RIP3-MLKL-mediated necroinflammation via Wnt3α/β-catenin/GSK-3β signaling in unilateral ureteral obstruction. International immunopharmacology. 2022;108:108868.
CrossRef - Snitow ME, Bhansali RS, Klein PS. Lithium and therapeutic targeting of GSK-3. Cells. 2021;10(2):255.
CrossRef - Khayachi A, Schorova L, Alda M, Rouleau G, Milnerwood A. Posttranslational modifications & lithium’s therapeutic effect—Potential biomarkers for clinical responses in psychiatric & neurodegenerative disorders. Neuroscience & Biobehavioral Reviews. 2021;127:424-445.
CrossRef - Rideout HJ, Chartier-Harlin M-C, Fell MJ, et al. The current state-of-the art of LRRK2-based biomarker assay development in PD. Frontiers in Neuroscience. 2020;14:865.
CrossRef - Chambers L, Seidler K, Barrow M. Nutritional entrainment of circadian rhythms under alignment and misalignment: A mechanistic review. Clinical nutrition ESPEN. 2022;51:50-71.
CrossRef - Lang AE, Espay AJ. Disease modification in PD: current approaches, challenges, and future considerations. Movement Disorders. 2018;33(5):660-677.
CrossRef - Deepak K, Roy PK, Das CK, Mukherjee B, Mandal M. Mitophagy at the crossroads of cancer development: exploring the role of mitophagy in tumor progression and therapy resistance. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2024:119752.
CrossRef - He S, Zhong S, Liu G, Yang J. Alpha-synuclein: The interplay of pathology, neuroinflammation, and environmental factors in PD. Neurodegenerative Diseases. 2020;20(2-3):55-64.
CrossRef - Peña-Zelayeta L, Delgado-Minjares K, Rojas MV, et al. Non-Motor Symptoms in PD. 2025;
- Hinault M-P, Farina-Henriquez-Cuendet A, Goloubinoff P. Molecular chaperones and associated cellular clearance mechanisms against toxic protein conformers in PD. Neurodegenerative Diseases. 2011;8(6):397-412.
CrossRef - Sharma R, Gupta V, Bora K, Dewangan HK. Targeting dopamine receptors with costunolide: a computational approach toward selective neuroprotection. J Neonatal Surg. 2025;14(31s):540-7.
CrossRef

