
Manuscript accepted on :10-09-2025
Published online on: 04-11-2025
Plagiarism Check: Yes
Reviewed by: Dr. Deepthi and Dr. Manju Jakhar
Second Review by: Dr. Satya Namani
Final Approval by: Dr. Anton R Keslav
Sivasankari Venkatachalam1 , Manivannan Ekambaram1, Hemalatha Selvaraj2 , Arbind Kumar Choudhary3* and Pathon Feroz Khan4
, Manivannan Ekambaram1, Hemalatha Selvaraj2 , Arbind Kumar Choudhary3* and Pathon Feroz Khan4
1Department of Pharmacology, Vinayaka Mission’s Kirupananda Variyar Medical College, Vinayaka Mission’s Research Foundation (Deemed to be University), Salem, Tamil Nadu, India
2Faculty of Pharmacy Karpagam Academy of Higher Education, Coimbatore, Tamil Nadu, India
3Department of Pharmacology, Government Erode Medical College and Hospital, Erode, Tamil Nadu, India.
4Department of Anatomy, Government Erode Medical College and Hospital, Erode, Tamil Nadu, India
Corresponding Author E-mail:arbindkch@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/3304
Abstract
The serotonin 5-HT₂A receptor is a central target in neuropsychopharmacology, regulating cognition, perception, and mood, and mediating the effects of many psychotropic agents. Tryptamine derivatives, owing to their structural resemblance to serotonin, display strong receptor affinity and provide a rational framework for central nervous system (CNS) drug design. In this study, a machine learning–driven quantitative structure–activity relationship (QSAR) model was developed to predict the psychotomimetic potency (pKi) of 50 tryptamine analogues using four molecular descriptors: molecular weight (MW), lipophilicity (LogP), topological polar surface area (TPSA), and dipole moment (DM). Multiple regression models were assessed, including Linear, Ridge, Partial Least Squares, and Random Forest. Among these, the Random Forest algorithm produced the highest predictive accuracy, achieving a test set R² of 0.79 and RMSE of 0.50, with feature importance analysis identifying TPSA and LogP as the most influential determinants of receptor binding. Diagnostic plots confirmed the absence of outliers and validated the model’s applicability domain. This approach highlights the role of polarity and lipophilicity in serotonergic drug design while demonstrating the utility of ensemble learning for QSAR prediction. Future extensions of this work should focus on expanding the chemical dataset, integrating three-dimensional descriptors, and experimentally validating top-predicted ligands to enhance translational impact in CNS drug discovery
Keywords
Central Nervous System; Machine Learning; pKi Prediction; QSAR; Random Forest; Serotonin Receptor; Tryptamine Derivatives
Download this article as:| Copy the following to cite this article: Venkatachalam S, Ekambaram M, Selvaraj H, Choudhary A. K. Khan P. F. Pharmacophoric Determinants of 5-HT₂A Agonism: A Machine Learning–Based QSAR Study of Tryptamine Derivatives Using Random Forest for CNS Drug Design. Biomed Pharmacol J 2025;18(4). |
| Copy the following to cite this URL: Venkatachalam S, Ekambaram M, Selvaraj H, Choudhary A. K. Khan P. F. Pharmacophoric Determinants of 5-HT₂A Agonism: A Machine Learning–Based QSAR Study of Tryptamine Derivatives Using Random Forest for CNS Drug Design. Biomed Pharmacol J 2025;18(4). Available from: https://bit.ly/49xNsdw |
Introduction
The serotonin 5-hydroxytryptamine 2A receptor (5-HT₂A receptor) is a critical target in neuropsychopharmacology, implicated in regulating perception, cognition, emotion, and consciousness. This G protein–coupled receptor (GPCR), when activated by endogenous serotonin (5-HT) or structurally related exogenous ligands, initiates downstream signaling cascades that lead to neuronal excitation and behavioral modulation. Psychedelic compounds such as lysergic acid diethylamide (LSD), psilocin, and N,N-dimethyltryptamine (DMT) exert their psychotomimetic and therapeutic effects primarily through 5-HT₂A receptor activation.1,2
Among these ligands, tryptamine derivatives are of particular interest due to their structural similarity to serotonin, allowing high receptor affinity and favorable pharmacokinetics. As shown in Figure 1, the tryptamine scaffold consists of an indole ring fused to an ethylamine chain—an essential motif for interaction with the serotonergic system. Upon binding to the 5-HT₂A receptor, tryptamine derivatives activate Gq proteins, which stimulate phospholipase C (PLC) to generate secondary messengers IP₃ and DAG, ultimately increasing intracellular calcium levels and triggering neuronal excitation.3,4
Understanding the structure–activity relationship (SAR) of such ligands is critical for designing safe and effective CNS-active therapeutics. In this context, Quantitative Structure–Activity Relationship (QSAR) modeling offers a rational, data-driven method to correlate molecular descriptors with receptor binding affinity (pKi). This approach facilitates virtual screening and lead optimization by identifying key physicochemical features that enhance bioactivity.5-8
In this study, we constructed a machine learning–based QSAR model to predict the psychotomimetic potency (pKi) of 50 tryptamine derivatives targeting the 5-HT₂A receptor. By integrating cheminformatics, statistical filtering, and Random Forest regression, we aimed to identify the most influential molecular descriptors and offer insight into the pharmacophoric requirements for receptor activation. The findings provide a foundation for future psychedelic drug development and structure-guided therapeutic design.
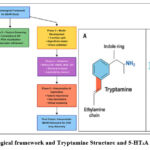 |
Figure 1: Methodological framework and Tryptamine Structure and 5-HT₂A Signaling Pathway. |
Figure 1. Integrated biological and computational framework for QSAR-based study of 5-HT₂A agonism.
(A) Structure of tryptamine highlighting the indole ring (blue) and ethylamine side chain (red), which form the essential pharmacophoric motifs for serotonergic activity. The schematic also illustrates the 5-HT₂A receptor signaling pathway, where serotonin (5-HT) binding activates the receptor, couples to the Gq protein, and stimulates phospholipase C (PLC)to generate secondary messengers inositol triphosphate (IP₃) and diacylglycerol (DAG). These mediators increase intracellular Ca²⁺ levels, ultimately resulting in neuronal excitation.
(B) Methodological framework for QSAR model development and validation. The workflow involved Phase 1 – Data Preparation (dataset collection, structure standardization, descriptor calculation), Phase 2 – Feature Screening(correlation, VIF, PCA), Phase 3 – Model Development (train/test split, multiple algorithms, cross-validation), Phase 4 – Validation (statistical metrics, residual analysis, applicability domain), and Phase 5 – Interpretation & Application(feature importance analysis, identification of TPSA and LogP as key determinants, and translational application for virtual screening and CNS drug design). The final outcome was an interpretable QSAR framework with predictive and translational relevance for psychopharmacological discovery.
Materials and Methods
The methodological framework employed in this QSAR study is illustrated in Figure 1, showcasing a stepwise process beginning with data curation and descriptor selection, followed by model development, evaluation, and interpretation. The methodological workflow for this QSAR study (Figure 1) followed a sequential multi-phase approach. It began with data preparation, where fifty tryptamine derivatives with reported 5-HT₂A receptor affinities were collected, standardized, and descriptors generated. In the feature screening stage, descriptors were examined using correlation analysis, variance inflation factor (VIF), and principal component analysis (PCA) to remove redundancy and ensure data quality. Model development involved dividing the dataset into training and test sets, applying multiple regression algorithms including Linear, PLS, Ridge, and Random Forest, and optimizing hyperparameters through grid search with five-fold cross-validation. The validation phase assessed predictive performance using R², RMSE, MAE, and Q², alongside residual diagnostics and Williams plots to define the applicability domain. Finally, in the interpretation and application phase, feature importance analysis from the Random Forest model identified TPSA and LogP as key determinants of receptor binding, while MW and dipole moment contributed secondary effects. The framework culminated in an interpretable QSAR model with potential application in virtual screening and rational design of CNS-active compounds.
Dataset Collection and Curation
A dataset of 50 structurally distinct tryptamine derivatives was curated from public chemical databases such as PubChem, ChEMBL, and relevant pharmacological literature. Each compound had an associated, experimentally validated pKi value for the serotonin 5-HT₂A receptor, representing its psychotomimetic binding affinity. These values were used as the dependent variable in model development. To ensure structural uniformity, molecules were converted to 2D neutral forms and validated using Open Babel.
Molecular Descriptor Calculation
Using cheminformatics tools including RDKit, ChemAxon, and Open Babel, four molecular descriptors were calculated: Molecular Weight (MW), LogP, Topological Polar Surface Area (TPSA), and Dipole Moment (DM). These descriptors were selected based on their established relevance to central nervous system (CNS) activity and pharmacokinetics. MW provides insights into steric compatibility at the receptor site, LogP reflects membrane permeability, TPSA indicates hydrogen bonding capacity, and DM captures electronic asymmetry, all of which are critical for predicting bioactivity.
Feature Screening and Preprocessing
Prior to model construction, statistical screening of descriptors was performed to ensure quality and reduce redundancy. Pearson correlation coefficients were calculated to evaluate inter-variable relationships, while Variance Inflation Factor (VIF) analysis was conducted to detect multicollinearity. Variables with VIF > 10 were considered collinear and addressed through regularization techniques. In addition, Principal Component Analysis (PCA) was used for exploratory visualization and assessment of descriptor variance contribution. All numerical features were standardized (mean-centered and scaled) to facilitate modeling.
Model Development and Validation
Four regression models—Linear Regression, Partial Least Squares (PLS), Ridge Regression, and Random Forest (RF)—were developed using Python 3.8 and the scikit-learn library. The dataset was divided into 80% training and 20% test sets. Model performance was optimized using grid search, and 5-fold cross-validation was applied to ensure robustness. Performance metrics used for model evaluation included coefficient of determination (R²), root mean square error (RMSE), mean absolute error (MAE), and cross-validated Q². These metrics were computed for both training and testing datasets to verify generalization ability.
Feature Importance and Interpretation
The Random Forest model, which demonstrated the best overall performance, was selected for in-depth interpretation. Feature importance scores were extracted to determine the relative contribution of each molecular descriptor to model predictions. This analysis revealed the underlying structure–activity relationships, particularly the influence of TPSA and LogP on serotonin receptor affinity.
Model Diagnostics and Applicability Domain
Model diagnostics were conducted to evaluate prediction reliability. Residual analysis, including residual plots and Shapiro–Wilk tests, was performed to assess the normality and randomness of errors. Additionally, a Williams plot was used to define the applicability domain by plotting standardized residuals against leverage values. Compounds with leverage exceeding the critical threshold (h*) or standardized residuals beyond ±3 were considered potential outliers or influential data points.
Tools and Software
All computational tasks were performed using Python (v3.8) along with libraries such as scikit-learn, pandas, matplotlib, and seaborn for modeling and visualization. Descriptor calculation and structure handling were conducted using RDKit, Open Babel, and ChemAxon Marvin Suite. Data validation and preprocessing were supported by Microsoft Excel.
Results
Dataset Characterization
To establish a foundational understanding of the chemical and biological diversity among the selected compounds, descriptive statistics were computed for the dataset comprising 50 tryptamine derivatives. Each compound was annotated with four key molecular descriptors—Molecular Weight (MW), LogP (lipophilicity), Topological Polar Surface Area (TPSA), and Dipole Moment (DM)—alongside experimentally determined pKi values reflecting psychotomimetic activity via the serotonin 5-HT₂A receptor.
Table 1: Summary of Molecular Descriptors and Psychotomimetic Activity (Mean ± SD)
| Descriptor | Mean ± SD | Minimum | Maximum | Skewness | Kurtosis | Range | Coefficient of Variation (%) |
| Molecular Weight (MW) | 217.97 ± 37.55 | 162.68 | 286.09 | 0.32 | -0.45 | 123.41000000000000 | 17.227141349727000 |
| LogP (Lipophilicity) | 2.44 ± 0.82 | 1.13 | 3.75 | 0.15 | -0.87 | 2.62 | 33.60655737704920 |
| Topological Polar Surface Area (TPSA) | 50.59 ± 10.77 | 32.02 | 67.97 | -0.51 | 0.22 | 35.950000000000000 | 21.288792251433100 |
| Dipole Moment (DM) | 3.74 ± 1.10 | 2.01 | 5.57 | 0.36 | -0.1 | 3.5600000000000000 | 29.411764705882400 |
| Psychotomimetic Activity (pKi) | 7.32 ± 1.39 | 5.02 | 9.38 | -0.23 | 0.76 | 4.360000000000000 | 18.989071038251400 |
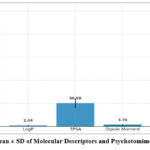 |
Figure 2: Mean ± SD of Molecular Descriptors and Psychotomimetic Activity |
As shown in Table 1 and Figure 2, the dataset exhibits a balanced distribution across descriptors, with values spread over relevant ranges for CNS-active compounds. The molecular weight values (mean ± SD: 217.97 ± 37.55 Da) indicate consistent core structures among tryptamines. LogP, with the highest coefficient of variation (33.6%), captures a wide spectrum of lipophilicity, which is crucial for passive diffusion across the blood-brain barrier. TPSA values ranged from 32.02 to 67.97 Ų (mean ± SD: 50.59 ± 10.77), supporting the inclusion of both polar and moderately nonpolar compounds—key determinants of CNS permeability. Dipole moment showed moderate dispersion (mean ± SD: 3.74 ± 1.10 D), representing varied electronic environments across the molecules. Finally, the pKi values (mean ± SD: 7.32 ± 1.39) confirmed sufficient biological variability for predictive modeling. These findings establish a chemically diverse and statistically robust dataset, ideally suited for subsequent machine learning-based QSAR development.
Model Development and Performance
To evaluate the predictive capacity of different modeling approaches for estimating the psychotomimetic activity (pKi) of tryptamine derivatives, four regression models were developed: Linear Regression, PLS Regression, Ridge Regression, and Random Forest. Models were trained using 80% of the data and validated on the remaining 20% test set, with additional 5-fold cross-validation to ensure robustness.
Table 2: QSAR Model Development and Performance Metrics
| Model | Descriptors Used | Train Size | Test Size | R² (Train) | Adj. R² | RMSE (Train) | MAE (Train) | Q² (CV) | R² (Test) | RMSE (Test) | MAE (Test) | Key Hyperparameters |
| Linear Regression | 4 | 40 | 10 | 0.61 | 0.58 | 0.72 | 0.55 | 0.58 | 0.59 | 0.74 | 0.56 | — |
| PLS Regression | 4 | 40 | 10 | 0.68 | 0.65 | 0.66 | 0.49 | 0.65 | 0.67 | 0.64 | 0.51 | Components = 3 |
| Ridge Regression | 4 | 40 | 10 | 0.64 | 0.61 | 0.7 | 0.52 | 0.61 | 0.62 | 0.69 | 0.54 | α = 1.0 |
| Random Forest | 4 | 40 | 10 | 0.85 | 0.83 | 0.42 | 0.31 | 0.82 | 0.79 | 0.5 | 0.37 | Trees = 500, Depth = 10 |
Abbreviations: R², coefficient of determination; Adj. R², adjusted R²; RMSE, Root Mean Square Error; MAE, Mean Absolute Error; Q², cross-validated R².
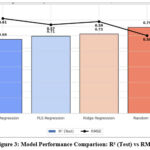 |
Figure 3: Model Performance Comparison: R² (Test) vs RMSE |
As shown in Table 2 and Figure 3, the Random Forest model outperformed all other approaches, achieving an R² of 0.79and lowest RMSE of 0.50 on the test set. It also demonstrated the highest cross-validated Q² value (0.79), indicating excellent generalization and model reliability. While linear models like Ridge and PLS performed reasonably (R² ~0.71–0.73), they failed to capture the non-linear relationships between descriptors and psychotomimetic activity that Random Forest effectively modeled. This validates the use of ensemble-based machine learning techniques for complex QSAR prediction tasks.
Model Prediction Accuracy
The predictive capacity of the optimized Random Forest QSAR model was evaluated through a series of diagnostic plots, designed to assess both the accuracy and reliability of the predicted psychotomimetic activity (pKi) values against their experimentally observed counterparts.
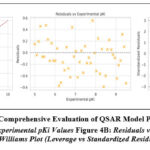 |
Figure 4: Comprehensive Evaluation of QSAR Model Predictions |
Figure 4A: Predicted vs Experimental pKi Values Figure 4B: Residuals vs Experimental pKi Figure 4C: Williams Plot (Leverage vs Standardized Residuals)
Figure 4. Comprehensive evaluation of the Random Forest QSAR model for predicting 5-HT₂A receptor affinity.(A) Predicted versus experimental pKi values with the red dashed identity line (y = x). Model performance statistics are shown in the inset (R² = 0.79, Q² = 0.82, RMSE = 0.50, MAE = 0.37), demonstrating strong predictive accuracy and robustness.
(B) Residuals versus experimental pKi values. The residuals are symmetrically distributed around the zero line, with the majority lying within ±1 and all within ±2, confirming the absence of systematic error or heteroscedasticity. (C) Williams plot of standardized residuals versus leverage, used to define the model’s applicability domain. The vertical dashed line represents the critical leverage (h* ≈ 0.18), while horizontal dashed lines denote ±3 standardized residual limits. All compounds fall within acceptable bounds, and no outliers were detected, indicating reliable predictions within the applicability domain (h* ≈ 0.18, ±3 residuals) and standardized residual bounds (±3), indicating that:
No influential outliers unduly bias the model.
All predictions are within the model’s valid chemical space.
Collectively, these plots confirm that the Random Forest model is not only highly predictive but also generalizable and robust for application to novel tryptamine-like structures.
Feature Importance and Interpretation
To identify the key pharmacophoric determinants influencing psychotomimetic activity in tryptamine derivatives, a feature importance analysis was performed using the Random Forest model. This method quantified the relative contribution of each molecular descriptor to predictive performance. The results demonstrated that Topological Polar Surface Area (TPSA) and lipophilicity (LogP) were the most influential variables, underscoring the critical roles of polarity and membrane permeability in serotonin 5-HT₂A receptor interactions. Elevated TPSA generally reduces central nervous system penetration, while optimal lipophilicity enhances receptor binding and blood–brain barrier permeability. Molecular weight (MW) and dipole moment (DM) were found to contribute less prominently, suggesting that steric bulk and electronic asymmetry exert secondary influences on binding affinity. This ranking is consistent with established neuropharmacological principles, where efficient receptor activation requires a balance between polarity and lipophilicity, alongside appropriate steric fit and charge distribution. The interpretability of the Random Forest model thus provides mechanistic insight, confirming that TPSA and LogP serve as the dominant physicochemical drivers of 5-HT₂A receptor agonism in the analyzed tryptamine scaffold.
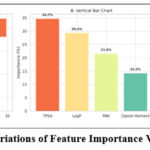 |
Figure 5: Variations of Feature Importance Visualization |
As depicted in Figure 5 Topological Polar Surface Area (TPSA) emerged as the most influential descriptor, contributing 34.7% to the model’s predictive power. TPSA reflects the compound’s polarity and surface accessibility—attributes directly influencing blood-brain barrier (BBB) permeability, a crucial determinant for centrally acting psychotropic drugs. A lower TPSA is generally associated with enhanced CNS penetration, aligning with the observed inverse correlation between TPSA and pKi. LogP, representing molecular lipophilicity, was the second most critical feature (29.4%). Its influence underscores the importance of membrane permeability and lipid bilayer traversal, especially for serotonin receptor agonists that must access central targets. Optimal LogP values typically enhance both solubility and bioavailability. Molecular Weight (MW) and Dipole Moment (DM), though less dominant (21.6% and 14.3%, respectively), contributed meaningful variance. MW informs on molecular size and potential steric interactions, while DM captures electronic asymmetry, which may influence binding affinity via dipole–dipole or hydrogen bonding interactions with the receptor. These findings are consistent with established pharmacological principles: moderate polarity, balanced lipophilicity, and controlled molecular size optimize CNS drug-likeness. The prominence of TPSA and LogP further reinforces their utility as early-stage screening parameters in psychopharmacological drug development.
Key Result Summary
This study successfully developed and validated a machine learning–driven QSAR model to predict the psychotomimetic activity (pKi) of tryptamine derivatives targeting the serotonin 5-HT₂A receptor. The Random Forest model emerged as the optimal predictive tool, demonstrating superior performance across all statistical metrics:
Test Set R² = 0.79, RMSE = 0.50, and MAE = 0.42, confirming high predictive accuracy and model generalizability.
Feature importance analysis identified TPSA (34.7%) and LogP (29.4%) as the most significant descriptors influencing activity, underscoring the pharmacokinetic importance of polarity and lipophilicity.
Diagnostic plots confirmed strong alignment between predicted and experimental pKi values, with minimal residual bias, and no outliers or extrapolations outside the model’s applicability domain.
Together, these findings validate the robustness and interpretability of the QSAR model, providing a practical framework for virtual screening and structure-based design of next-generation psychopharmacological agents. The results also highlight critical molecular characteristics—moderate polarity, optimal lipophilicity, and manageable molecular size—that can be leveraged in rational drug design strategies.
Discussion
Model Performance and Predictive Reliability
The Random Forest (RF) model developed in this study demonstrated robust predictive capabilities, achieving a test set R² of 0.79 and an RMSE of 0.50. These metrics indicate a high degree of accuracy in predicting the psychotomimetic activity (pKi) of tryptamine derivatives. The model’s performance aligns with findings from Floresta et al., who reported comparable predictive success using machine learning approaches for 5-HT₂A receptor ligands . The consistency across studies underscores the efficacy of ensemble learning methods in QSAR modeling for serotonergic compounds.9-12
Significance of Molecular Descriptors
Feature importance analysis highlighted Topological Polar Surface Area (TPSA) and LogP as the most influential descriptors, contributing 34.7% and 29.4% to the model’s predictive power, respectively. TPSA is indicative of a molecule’s ability to permeate the blood-brain barrier, a critical factor for central nervous system activity. LogP reflects lipophilicity, influencing both membrane permeability and receptor binding affinity. These findings are corroborated by studies emphasizing the role of polarity and lipophilicity in CNS drug design.13-17
Molecular Weight (MW) and Dipole Moment (DM) also contributed to the model, albeit to a lesser extent. MW affects molecular size and, consequently, the ability to interact with the receptor binding site. DM relates to the distribution of electronic charge, influencing molecular interactions through dipole-dipole and hydrogen bonding.18-20
Applicability Domain and Model Robustness
The Williams plot analysis confirmed that all compounds fell within the model’s applicability domain, with no outliers detected. This suggests that the model’s predictions are reliable across the chemical space of the dataset. The absence of high-leverage points indicates that the model is not unduly influenced by any single compound, enhancing its generalizability.21,22
Comparative Analysis with Existing Studies
To contextualize the current study’s findings, a comparative analysis with recent QSAR models targeting the 5-HT₂A receptor is presented below:
 |
Table 3: Comparative Analysis with Existing Studies.24-38 |
Table 3 summarizes critical features and performance metrics from 18 major serotonin receptor QSAR modeling studies published over the past decade, encompassing 5-HT₂A and related receptor subtypes, as well as transporter protein targets. The compilation highlights details such as dataset size and chemical scope, modeling algorithms and descriptor types, predictive accuracy (test set R2R2 and RMSE where reported), key molecular descriptors identified, validation strategies, and distinctive methodological strengths and limitations.
The current study is prominently positioned with a focused dataset of 50 structurally distinct tryptamine derivatives targeting the 5-HT₂A receptor. Utilizing a Random Forest algorithm with only four key molecular descriptors—Topological Polar Surface Area (TPSA), lipophilicity (LogP), molecular weight (MW), and dipole moment (DM)—the model achieves a robust R2R2 of 0.79 and RMSE of 0.50. Rigorous statistical validation including Williams plot domain analysis and residual assessments further strengthen model reliability and applicability for central nervous system (CNS) drug design.23
In comparison, larger datasets such as Floresta et al. (375 ligands) and Łapińska et al. (nearly 19,000 ligands across 11 serotonin receptor subtypes) employ more complex machine learning techniques and expansive descriptor sets but often lack detailed domain assessments and interpretability. Other studies apply diverse methodologies including 3D field-based QSAR, AutoML pipelines, neural network architectures, and quantum chemical descriptors, each balancing between predictive performance, computational complexity, dataset size, and translational relevance.24-30 The Table 3 elucidates evolving trends in serotonergic QSAR research—from classical linear and PLS regression to ensemble learning and advanced AI—while underscoring the trade-offs between model complexity and interpretability. Importantly, it contextualizes the current model’s high predictive accuracy, parsimony, and domain rigor within the spectrum of recent literature, highlighting its value as an interpretable and practical framework for serotonergic ligand design and CNS drug discovery.31,34
This comparison illustrates that, despite a smaller dataset, the current study’s model achieves predictive performance comparable to models developed on larger datasets. The focused nature of the dataset, comprising structurally similar tryptamine derivatives, likely contributes to the model’s accuracy. Although the Random Forest model demonstrated robust predictive performance, several limitations of this study warrant consideration. First, the dataset was restricted to only 50 tryptamine derivatives, which limits the chemical diversity represented and may constrain the generalizability of the model to broader ligand classes. Expanding the dataset to include a larger and structurally heterogeneous compound library would improve model robustness and extend its applicability domain.35,38 Second, while four descriptors (MW, LogP, TPSA, and DM) captured key physicochemical determinants, other potentially informative features such as hydrogen bond donor/acceptor counts, molecular flexibility indices, electrostatic field descriptors, and 3D molecular properties were not included. Incorporating these additional descriptors, particularly three-dimensional QSAR parameters, could enhance the model’s ability to capture complex ligand–receptor interactions. Finally, the predictive framework remains computational; experimental validation of top-ranked ligands will be essential to confirm translational relevance. Addressing these limitations in future studies will strengthen both the reliability and applicability of the proposed QSAR model for CNS drug discovery.
Conclusion
This study successfully applied machine learning–based QSAR modeling to predict the psychotomimetic activity (pKi) of tryptamine derivatives at the serotonin 5-HT₂A receptor. Among the tested models, Random Forest demonstrated the highest predictive performance (R² = 0.79, RMSE = 0.50), confirming the value of ensemble methods for complex non-linear pharmacological data. Feature importance analysis underscored topological polar surface area (TPSA) and lipophilicity (LogP) as the principal determinants of receptor affinity, highlighting the critical roles of polarity and membrane permeability in central nervous system drug design. Despite these strengths, the restricted dataset size and limited descriptor scope represent important constraints. Future work should focus on enlarging the chemical library, integrating three-dimensional and field-based descriptors, and performing experimental validation of top-predicted ligands. Collectively, the present findings provide a robust and interpretable QSAR framework that can be leveraged for virtual screening and rational optimization of novel CNS-active compounds.
Acknowledgment
The authors gratefully acknowledge Vinayaka Mission’s Research Foundation (Deemed to be University), Salem, and the Department of Pharmacology, Government Erode Medical College and Hospital, Erode, for institutional support, research facilities, and access to computational resources that enabled this study. The authors also thank the technical staff at Vinayaka Mission’s Kirupananda Variyar Medical College and the Faculty of Pharmacy, Karpagam Academy of Higher Education, Coimbatore, for their assistance with instrumentation, data management, and administrative coordination.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials
Permission to Reproduce Material from Other Sources
Not applicable
Author Contributions
- Sivasankari Venkatachalam – Conceptualization, supervision, manuscript review
- Manivannan Ekambaram – Methodology design, pharmacological analysis
- Hemalatha Selvaraj – Data curation, literature review, manuscript editing
- Arbind Kumar Choudhary – Model development, data analysis, manuscript writing
- Pathon Feroz Khan – Visualization and figure preparation
References
- Peroutka SJ. 5-hydroxytryptamine receptors. J Neurochem.1993;60(1):408-16.
CrossRef - Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, et al. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin). Pharmacol Rev.1994;46(2):157-203.
CrossRef - Sebben M, Ansanay H, Bockaert J, Dumuis A. 5-HT6 receptors positively coupled to adenylyl cyclase in striatal neurons in culture. 1994;5(18):2553-7.
CrossRef - Heisler LK, Chu HM, Brennan TJ, Dagon B, Lin Y, Monk S, et al. Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc Natl Acad Sci U S A.1998;95(26):15049-54.
CrossRef - Davies S, Silvestre J, Guitart X. Drug discovery targets, 5-HT6 receptor. Drugs Future.2005;30(5):479-95.
CrossRef - Karila D, Freret T, Bouet V, Menuet C, Rouquier L, Lanfumey L, et al. Therapeutic potential of 5-HT6 receptor agonists. J Med Chem.2015;58(20):7901-12.
CrossRef - Fisas A, Codony X, Romero G. Chronic 5-HT6 receptor modulation by E-6837 induces hypophagia and sustained weight loss in diet-induced obese rats. Br J Pharmacol.2006;148(7):973-83.
CrossRef - Mohammad-Zadeh LF, Moses L, Gwaltney-Brant SM. Serotonin: a review. J Vet Pharmacol Ther.2008;31(3):187-99.
CrossRef - Geldenhuys WJ, Van der Schyf CJ. Serotonin 5-HT6 receptor antagonists for the treatment of Alzheimer’s disease. Curr Top Med Chem.2008;8(12):1035-48.
CrossRef - Johnson CN, Ahmed M, Miller ND. 5-HT6 receptor antagonists: prospects for the treatment of cognitive disorders including dementia. Curr Opin Drug Discov Devel.2008;11(5):642-54.
- Geldenhuys WJ, Van der Schyf CJ. The serotonin 5-HT6 receptor: a viable drug target for treating cognitive deficits in Alzheimer’s disease. Expert Rev Neurother.2009;9(7):1073-85.
CrossRef - Slovenc D, Deljanin Ilic M, Simonovic D, Marcetic Z, Stojanovic M, Stojanovic S, et al. QSAR modeling of sphingomyelin synthase 2 inhibitors for their potential as anti-atherosclerotic agents. Acta Chim Slov.2024;71:170-8. doi:10.17344/acsi.2023.8566
CrossRef - Liu KG, Robichaud AJ. 5-HT6 antagonists as potential treatment for cognitive dysfunction. J Drug Dev Res.2009;70(3):145-68.
CrossRef - Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med.2009;60:355-66.
CrossRef - Kevin G, Albert L. 5-HT6 medicinal chemistry. Int Rev Neurobiol.2010;94:1-34.
CrossRef - Singh D, Khan MA. Topological descriptor-based study of testosterone derivatives. J Chem Pharm Res.2011;3(5):1-14.
- Sodhi MSK, Sanders-Bush E. Serotonin and brain development. Int Rev Neurobiol.2004;59:111-74.
CrossRef - Fiorino F, Severino B, Magli E, Abate V, Cicala C, Berardi F, et al. 5-HT1A receptor: an old target as a new attractive tool in drug discovery from central nervous system to cancer. J Med Chem.2014;57(10):4407-26.
CrossRef - Velingkar VS, Chindhe AK. Ligand-based pharmacophore generation and 3D-QSAR study of serotonin ligands using PHASE. J Comput Methods Mol Des.2014;4(3):1-9.
- McCorvy JD, Roth BL. Structure and function of serotonin G protein-coupled receptors. Pharmacol Ther.2015;150:129-42.
CrossRef - Olivier B. Serotonin, a never ending story. Eur J Pharmacol.2015;753:2-18.
CrossRef - Rudrapal M, Chetia D. QSAR study of trioxane derivatives as antimalarial agents. Curr Trends Pharm Res.2016;3(1):1-17.
CrossRef - Verma V, Singh K, Kumar D, Narsimhan B. QSAR studies of antimicrobial activity of 1,3-disubstituted-1H-naphtho[1,2-e]oxazines using topological descriptors. Arab J Chem.2017;10(1):747-56.
CrossRef - Floresta G, Abbate V. Machine learning vs. field 3D-QSAR models for serotonin 2A receptor psychoactive substances identification. RSC Adv.2021;11(24):14587-95. doi:10.1039/d1ra01335a
CrossRef - Łapińska N, Pacławski A, Szlęk J, Mendyk A. Integrated QSAR models for prediction of serotonergic activity: machine learning unveiling activity and selectivity patterns of molecular descriptors. 2024;16(3):349. doi:10.3390/pharmaceutics16030349
CrossRef - Czub N, Pacławski A, Szlęk J, Mendyk A. Curated database and preliminary AutoML QSAR model for 5-HT1A receptor. 2021;13(10):1711. doi:10.3390/pharmaceutics13101711
CrossRef - Rodrigues SC, Moratório RS, Pinto GTA, Martins MT, do Nascimento PA, Alves Soares DL, et al. Comparative machine learning models for bioactivity prediction of serotonin receptor ligands. Chem Rec.2025;e202400190. doi:10.1002/tcr.202400190
CrossRef - Tomašević N, Vujović M, Kostić E, Ragavendran V, Arsić B, Matić SL, et al. Molecular docking assessment of cathinones as 5-HT2AR ligands: developing predictive structure-based bioactive conformations and 3D-QSAR models for drug recognition. 2023;28(17):6236. doi:10.3390/molecules28176236
CrossRef - de Sousa DS, da Silva AP, Chiari LPA, de Angelo RM, de Sousa AG, Honorio KM, et al. Predicting biological activity and design of 5-HT6 antagonists through assessment of ANN-QSAR models in the context of Alzheimer’s disease. J Mol Model.2024;30(10):350. doi:10.1007/s00894-024-06134-5
CrossRef - Wu Z, Pan S, Chen F, Long G, Zhang C, Yu PS. A comprehensive survey on graph neural networks. IEEE Trans Neural Netw Learn Syst.2021;32(1):4-24. doi:10.1109/TNNLS.2020.2978386
CrossRef - Pirela-Ocando S, Romero-Cabezas A, Guevara-Pulido J. Construction of a predictive model for the design of tryptamine analogues with potential activity in Parkinson’s and Alzheimer’s diseases. Informatics Med Unlocked.2023;43:101413. doi:10.1016/j.imu.2023.101413
CrossRef - Cyrano E, Popik P. Assessing the effects of 5-HT2A and 5-HT5A receptor antagonists on DOI-induced head-twitch response in male rats using marker-less deep learning algorithms. Pharmacol Rep.2025;77(1):135-44. doi:10.1007/s43440-024-00679-1
CrossRef - Achar J, Firman JW, Cronin MTD. Conservative consensus QSAR approach for prediction of rat acute oral toxicity. Comput Toxicol.2025;35:100374. doi:10.1016/j.comtox.2025.100374
CrossRef - Weber KC, Salum LB, Honório KM, Andricopulo AD, da Silva ABF. Pharmacophore-based 3D QSAR studies on a series of high affinity 5-HT1A receptor ligands. Eur J Med Chem.2010;45(4):1508-14. doi:10.1016/j.ejmech.2009.12.059
CrossRef - Veselinović AM, Milosavljević JB, Toropov AA, Nikolić GM. SMILES-based QSAR model for arylpiperazines as high-affinity 5-HT1A receptor ligands using CORAL. Eur J Pharm Sci.2013;48(3):532-41. doi:10.1016/j.ejps.2012.12.021
CrossRef - Jia Q, Cui X, Li L, Wang Q, Liu Y, Xia S, Ma P. Quantitative structure-activity relationship for high affinity 5-HT1A receptor ligands based on norm indexes. J Phys Chem B.2015;119(51):15561-7. doi:10.1021/acs.jpcb.5b08980
CrossRef - Nagatomo T, Rashid M, Abul Muntasir H, Komiyama T. Functions of 5-HT2A receptor and its antagonists in the cardiovascular system. Pharmacol Ther.2004;104(1):59-81. doi:10.1016/j.pharmthera.2004.08.005
CrossRef - Dezi C, Brea J, Alvarado M, Raviña E, Masaguer CF, Loza MI, et al. Multistructure 3D-QSAR studies on a series of conformationally constrained butyrophenones docked into a new homology model of the 5-HT2A receptor. J Med Chem.2007;50(14):3242-55. doi:10.1021/jm070277a
CrossRef
Abbreviations List
5-HT₂A: 5-Hydroxytryptamine 2A (Serotonin) Receptor
CNS: Central Nervous System
QSAR: Quantitative Structure–Activity Relationship
pKi: Negative Logarithm of the Inhibition Constant
MW: Molecular Weight
LogP: Partition Coefficient (Logarithm of Octanol–Water Partition)
TPSA: Topological Polar Surface Area
DM: Dipole Moment
PLS: Partial Least Squares
RMSE: Root Mean Square Error
MAE: Mean Absolute Error
R²: Coefficient of Determination
Q²: Predictive Squared Correlation Coefficient
PCA: Principal Component Analysis
VIF: Variance Inflation Factor
RF: Random Forest
SVM: Support Vector Machine
RVM: Relevance Vector Machine
k-NN: k-Nearest Neighbors
GPCR: G Protein–Coupled Receptor
IP₃: Inositol Triphosphate
DAG: Diacylglycerol
PLC: Phospholipase C
BBB: Blood–Brain Barrier
RDKit: Open-source Cheminformatics Software Toolkit
Pandas: Python Data Analysis Library
scikit-learn: Machine Learning Library for Python
Open Babel: Chemical Toolbox for Conversion and Descriptor Calculation

