
Manuscript accepted on :23-07-2025
Published online on: 25-08-2025
Plagiarism Check: Yes
Reviewed by: Dr. Kasthuri NMK
Second Review by: Dr. Medvedev and Dr. Swati Jogdand
Final Approval by: Dr. Anton R Keslav
Magesh Mohan1 , Renukadevi Jeyavelkumaran2*, Vijayakumar Balakrishnan3and Sanjay Valliappan2
, Renukadevi Jeyavelkumaran2*, Vijayakumar Balakrishnan3and Sanjay Valliappan2
1Department of Pharmaceutical Chemistry, Saveetha College of Pharmacy, SIMATS, Chennai, Tamil nadu, India
2Department of Pharmaceutics, Saveetha College of Pharmacy, SIMATS, Chennai, Tamil nadu, India.
3Department of Pharmaceutical Chemistry, Grace College of Pharmacy, Kerala University of Health Sciences, Palakkad, Kerala, India
Corresponding author Email id - renukadevij.scop@saveetha.com
DOI : https://dx.doi.org/10.13005/bpj/3238
Abstract
Cofilin is crucial for the maintenance of neuronal architecture, synaptic plasticity, and intracellular transport. The dimerization of cofilin under oxidative stress contributes to the formation of cofilin-actin rods, which severely interfere with synaptic signaling, axonal transport, and intracellular trafficking. Such pathological changes are hallmarks of various neurodegenerative diseases. This study aimed at the identification of small-molecule inhibitors targeting cofilin dimerization, which could restore its monomeric, actin-severing state. Eighteen candidate compounds were screened using molecular docking and molecular dynamics simulations to assess their potential to disrupt the dimerization interface. Asiaticoside, Actinorhodin, and Granatin A demonstrated the most favorable binding affinities, with Asiaticoside emerging as the strongest inhibitor (-7.3 kcal/mol). Derived from Bacopa monnieri (Brahmi), Asiaticoside displayed stable binding to critical dimerization residues, as confirmed by molecular dynamics simulations. These simulations showed a stable protein-ligand complex with minimal structural fluctuations, a consistent hydrogen bond network, and strong electrostatic and hydrophobic interactions. This work highlights the therapeutic potential for targeting cofilin dimerization to restore actin filament dynamics and ameliorate neurodegenerative disease-associated cytoskeletal dysfunction. The overall findings suggest Asiaticoside to be a promising lead compound, with experimental validation and optimization being necessary to enhance the translational application in the treatment of neurodegenerative disorders.
Keywords
Asiaticoside; Cofilin dimer inhibitors; Cognitive decline; GROMACS; MD simulation
Download this article as:| Copy the following to cite this article: Mohan M, Jeyavelkumaran R, Balakrishnan V, Valliappan S. Molecular Modelling Study of Cofilin Dimer Inhibitors in Cognitive Decline. Biomed Pharmacol J 2025;18(3). |
| Copy the following to cite this URL: Mohan M, Jeyavelkumaran R, Balakrishnan V, Valliappan S. Molecular Modelling Study of Cofilin Dimer Inhibitors in Cognitive Decline. Biomed Pharmacol J 2025;18(3). Available from: https://bit.ly/47vD5pV |
Introduction
Cognitive decline is manifested as memory impairment, reduced executive function, and reduced cognitive abilities. This usually involves synaptic dysfunction and disrupted neuronal signaling in association with cytoskeletal instability.1 Given the importance of the actin cytoskeleton to maintain neuronal structure, synaptic plasticity, and intracellular transport, disruptions in actin filament dynamics are particularly vulnerable to effects.2 The emerging evidence of cofilin as a critical actin-binding protein plays a central role in these processes and the pathobiology of most neurodegenerative diseases. Severing and depolymerizing actin filaments are essential for their turnover, an upstream process needed for cellular motility, intracellular transport, and synaptic plasticity.3 Its function is highly dependent on its structural state with its existence in monomeric and dimeric forms. Cofilin, however, forms intermolecular disulfide bonds under oxidative stress, or in pathological conditions, to form a dimeric structure with a functional switch transforming it from a severing protein into a bundling agent.4 This process is responsible for the aggregation of actin filaments into cofilin-actin rods with serious consequences for normal cellular processes.4 Cofilin is extremely responsible for affecting synaptic signaling, axonal transport, and intracellular trafficking.5 A characteristic feature of neurodegenerative diseases such as Alzheimer’s disease (AD) is the accumulation of cofilin–actin rods, and levels of these rods correlate with synaptic deficits and progression of cognitive decline.6 Oxidative stress is a common feature in neurodegeneration and is tightly linked to the monomeric to dimeric transition of cofilin.7 Thiol modifications in cofilin by reactive oxygen species (ROS) may also lead to dimer formation. In this oxidative environment, the formation of cofilin actin rods is exacerbated and the pool of free monomeric cofilin is decreased leading to the onset of cognitive declines.4 These pathological implications suggest inhibition of cofilin dimerization appears to be a promising therapeutic strategy to treat cognitive decline. Natural compounds showed great potential as particularly good candidates for neuroprotection due to their neuroprotective properties, bioavailability, and low toxicity. Insights into binding affinities and key interactions from molecular docking, stability, and behavior of the cofilin inhibitor complexes under physiological conditions will provide an insight into the understanding of cofilin’s role as inhibitors in neurodegenerative diseases thus in the development of future drug-lead interventions in drug discovery. This work aims to assess the natural product-based cofilin inhibitors using molecular modeling studies.
Materials and Methods
The human Cofilin monomer and dimer structures were modeled using AlphaFold, and the dimer interface was analyzed using the Arpeggio webserver to identify residues involved in protein-protein interactions. Key residues contributing to dimerization, including ALA-105, ALA-109, ALA-118, ALA-123, ALA-137, ASN-138, ASP-122, CYS-80, GLN-136, GLU-107, GLU-142, LEU-111, LYS-112, LYS-114, LYS-121, LYS-125, LYS-126, MET-115, PHE-103, PRO-106, PRO-110, SER-108, SER-113, SER-119, SER-120, TRP-104, TYR-82, and TYR-117, were identified. The monomer structure was validated using SAVESv6.1. Eighteen small molecules were selected to target the dimer interface, including Asiaticoside, Actinorhodin, Granatin A, Asiatic Acid, Madecassic Acid, Deoxycholic Acid, Urolithin A, Indole-3-acetic acid, 3-Indolepropionic acid, Menadione, Phenylacetylglutamine, Salicylic Acid, Lipoteichoic Acid, Sphinganine, Gamma-Aminobutyric Acid, Lactate, Butanoate, and Propionate ion. Ligand structures were retrieved from the PubChem database and prepared for docking using AutoDockTools. Polar hydrogens were added, and Gasteiger charges were assigned to the ligands. The protein was prepared by retaining it in a rigid configuration while allowing full flexibility to the ligands.
Molecular Docking
Molecular docking was performed using AutoDock, with the grid box centered on the identified dimer interface. The grid center coordinates were center_x = -7.806, center_y = -11.253, center_z = 1.399 and size_x = 41,size_y = 34, size_z = 29.8 The exhaustiveness parameter was set to 24 to ensure comprehensive sampling. Docking results were evaluated based on binding energy scores, with the top-ranking compounds subjected to further interaction analysis. The spatial orientation of the ligands and their interactions with key residues were visualized using PyMOL and Discovery Studio.9 The binding energies and interaction profiles of the top three compounds were extensively analyzed to determine their potential to disrupt dimerization. These findings provide insights into the inhibitory mechanisms of these compounds and their potential therapeutic relevance.
Molecular Dynamics simulation
Molecular dynamics (MD) simulations were conducted using GROMACS 2023.1 to evaluate the stability and dynamics of the protein-ligand complex.10 The protein structure was prepared using the AMBER99SB-ILDN force field, and the SPC water model was selected for solvation. Initial preparation of the protein involved the gmx pdb2gmx command to generate the topology and position restraints files. The system was solvated with 12,962 water molecules, and electroneutrality was achieved by adding 23 sodium (Na⁺) and 22 chloride (Cl⁻) ions. Energy minimization was performed using the steepest descent algorithm for 1,000 steps, with an energy convergence criterion of 1,000 kJ/mol/nm.
The system was equilibrated in two stages: NVT (constant number of particles, volume, and temperature) for 1 ns and NPT (constant number of particles, pressure, and temperature) for 1 ns, both with position restraints applied to the protein and ligand. The V-rescale thermostat was used to maintain the temperature at 310.15 K, with separate coupling groups for the protein, ligand, and water/ions. During the NPT equilibration, the pressure was regulated at 1 bar using the Parrinello-Rahman barostat.
A 100 ns production MD simulation was performed with a time step of 2 fs, employing the Particle Mesh Ewald (PME) method for long-range electrostatic interactions and a 1.4 nm cutoff for short-range electrostatic and van der Waals interactions. The LINCS algorithm was applied to constrain all bonds, including those involving hydrogen atoms. Periodic boundary conditions (PBC) were applied in all three dimensions, and dispersion corrections were included for long-range van der Waals interactions. Trajectory data was saved every 10 ps in a compressed format, and energy and log files were updated at the same intervals. Post-simulation analyses focused on stability, conformational changes, and key interactions within the protein-ligand complex.
Free energy calculations were performed using the gmx_MMPBSA tool to determine the binding affinity and interaction dynamics. Both Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) and Molecular Mechanics Generalized Born Surface Area (MM-GBSA) methods were employed. Trajectory frames were selected every 100th frame from the MD simulation, covering frames 1 to 10,000, with the system maintained at 310.15 K. For the MM-GBSA calculations, the GB-OBC2 model (igb = 5) was used with a salt concentration of 0.150 M, a surface tension parameter of 0.0072, and internal and external dielectric constants set to 2.0 and 80.0, respectively. For MM-PBSA, a grid-based setup was employed with a fill ratio of 4.0 and similar dielectric constants and salt concentrations.
Residue-level energy decomposition analysis identified key residues involved in ligand binding. Residues within 6 Å of the ligand were included in this analysis, and energy contributions from electrostatic, van der Waals, and solvation components were calculated. The results provided insights into the molecular determinants of binding, highlighting critical residues in the binding pocket and their respective energy contributions. Outputs included the electrostatic (EEL), van der Waals (VDWAALS), and solvation-free energy terms, offering a detailed understanding of the binding mechanism and interaction energetics between the protein and ligand.
Results
Docking Results
The top three compounds identified from docking studies—Asiaticoside (-7.3 kcal/mol), Actinorhodin (-6.9 kcal/mol), and Granatin A (-6.8 kcal/mol)—showed the most favorable binding energies against the human Cofilin dimer interface (Table 1). These compounds were selected for further analysis to explore their binding modes and interactions with key residues at the dimerization interface
Table 1: Docking scores of compounds with cofilin protein
| File Name | Best Affinity |
| Propionate | -3.3 |
| Butyrate | -3.4 |
| Asiatic acid | -6.7 |
| Asiaticoside | -7.3 |
| GABA | -3.8 |
| Granatin A | -6.8 |
| Lipoteichoic acid | -4.3 |
| Deoxycholic acid | -6.4 |
| Salicylic acid | -4.6 |
| Indole 3 propionic Acid | -5.4 |
| Menadione | -5.3 |
| Actinorhodin | -6.9 |
| Urolithin A | -5.6 |
| Madecassic acid | -6.3 |
| Indole 3 acetic acid | -5.3 |
| Lactate | -3.4 |
| Sphinganine | -3.7 |
| Phenyacetyl glutamine | -5.7 |
Interaction Analysis
Asiaticoside exhibited the strongest binding affinity with a docking score of -7.3 kcal/mol. The analysis revealed conventional hydrogen bonds with LYS-114, ASN-138, and GLU-107, along with a pi-alkyl interaction with ALA-118. These interactions indicate a stable binding at the dimer interface, potentially interfering with the dimerization process as shown in FIG 1. The spatial alignment of Asiaticoside with key residues in the interaction interface suggests its ability to disrupt critical contacts necessary for dimer stability. Actinorhodin displayed a docking score of -6.9 kcal/mol, forming hydrogen bonds with LYS-121, ASP-122, and ASN-138, as well as salt bridges with LYS-114 and LYS-125. Hydrophobic interactions were observed with PHE-103, LEU-111, and ALA-118. These interactions suggest that Actinorhodin can effectively occupy the dimerization interface, disrupting both hydrophobic and electrostatic interactions critical for dimer formation.
Granatin A achieved a docking score of -6.8 kcal/mol. It formed hydrogen bonds with GLU-107, LYS-114, and ASN-138, as well as hydrophobic interactions with ALA-118 and LYS-114. Salt bridge formation with LYS-114 further stabilized its binding at the interface. The combination of strong hydrogen bonding and electrostatic interactions highlights Granatin A’s potential to interfere with dimerization by targeting multiple critical residues.
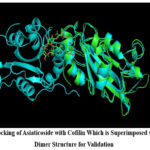 |
Figure 1: Docking of Asiaticoside with Cofilin Which is Superimposed with Cofilin Dimer Structure for ValidationClick here to view Figure |
Molecular dynamic simulation and Free energy Studies
The dynamics simulation results provide comprehensive insights into the stability and dynamics of the protein-ligand complex with Asiaticoside. The Root Mean Square Deviation (RMSD) graph (Fig 2.) shows the protein’s backbone stability over the 100 ns simulation, with values fluctuating between 0.1 nm and 0.25 nm and stabilizing around 0.15 nm after the equilibration phase. This indicates the protein maintains its structural integrity throughout the simulation without significant deviations.
The ligand RMSD (0.244 nm ± 0.025) suggests that Asiaticoside remains stably bound within the active site, undergoing minor positional adjustments, which reflect typical flexibility within the binding pocket (Fig 3). The minimum distance analysis confirms a stable interaction between the protein and ligand, with an average distance of 0.1659 nm (± 0.0111 nm), further corroborating the consistent positioning of Asiaticoside within the binding pocket. Minor fluctuations in this distance highlight the natural dynamics of the interaction without any significant dissociation events.
 |
Figure 2: Root Mean Square Deviation (Rmsd)Click here to view Figure |
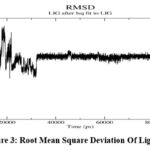 |
Figure 3: Root Mean Square Deviation of LigandClick here to view Figure |
The hydrogen bond analysis (Fig 4.) reveals that an average of 3.81 hydrogen bonds (± 1.20) are maintained between the protein and ligand throughout the simulation. Peaks in the hydrogen bond graph indicate transient increases in bonding, possibly due to ligand reorientation or interactions with additional residues, showcasing the dynamic yet stable nature of the interactions. The Root Mean Square Fluctuation (RMSF) analysis indicates low fluctuations (average RMSF: 0.130 nm ± 0.071) for most residues, with higher fluctuations observed in flexible regions such as the N- and C-termini and specific loops, shown in Fig 5. These regions are distant from the binding site, ensuring that Asiaticoside binding does not destabilize the protein’s core structure.
The Radius of Gyration (Rg) ) as shown in Fig 6 remains constant at 1.612 nm (± 0.009), indicating that Asiaticoside binding does not cause structural expansion or compaction, further affirming the compactness of the protein fold. Additionally, the Solvent Accessible Surface Area (SASA) as shown in Fig 7, fluctuates around an average of 97.95 nm² (± 1.95 nm²), suggesting consistent solvent exposure of the protein during the simulation. Minor variations in SASA reflect small conformational changes or adjustments due to the ligand, without significant disruption of the protein’s surface. Collectively, the RMSD, Rg, SASA, and minimum distance analyses confirm the overall stability of the protein-ligand complex throughout the simulation. Asiaticoside consistently interacts within the binding pocket, supported by its stable hydrogen bond network and minimal effects on the protein’s structural integrity. These findings highlight Asiaticoside’s potential as a strong inhibitor, effectively binding to the protein’s active site and maintaining stable interactions throughout the simulation.
 |
Figure 4: Hydrogen bond analysis of the protein-ligand complexClick here to view Figure |
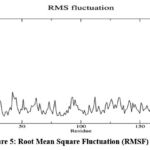 |
Figure 5: Root Mean Square Fluctuation (RMSF) analysisClick here to view Figure |
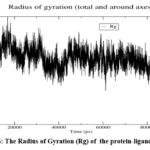 |
Figure 6: The Radius of Gyration (Rg) of the protein-ligand complexClick here to view Figure |
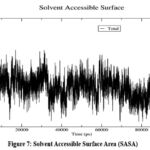 |
Figure 7: Solvent Accessible Surface Area (SASA) Click here to view Figure |
The results of the principal component analysis (PCA) reveal insights into the protein’s dynamic behavior during the simulation. The RMS fluctuation analysis ( Fig 8 )along the top five principal components highlights dominant motions in specific residues, particularly in flexible regions such as loops and termini. Significant peaks in eigenvectors 1 (vec 1) and 3 (vec 3) suggest that these components capture the most pronounced fluctuations, while lower fluctuations in vec 2, vec 4, and vec 5 indicate less substantial contributions to the overall flexibility.
The 2D projection of the trajectory ( Fig 9 ), which maps the motion along eigenvectors 1 and 5, demonstrates two distinct clusters, representing stable conformational states sampled during the simulation. The dense clustering within these regions reflects structural stability, while occasional transitions between clusters suggest minor shifts in the protein’s global conformation. This indicates that ligand binding induces significant yet stable conformational adjustments in the protein
.The eigenvector component analysis ( Fig 10) further reveals the anisotropic nature of the protein’s motion, with specific directional contributions from x and z components dominating in vec 1, and the y component contributing prominently in vec 3. These directional motions emphasize the dynamic nature of the protein-ligand interaction and the adaptability of the protein structure during binding. Collectively, the PCA results suggest that Asiaticoside binding stabilizes the protein in well-defined conformational states while inducing localized flexibility in critical regions. These observations underscore the ligand’s ability to modulate the protein’s conformational landscape, maintaining structural stability while enabling dynamic adjustments necessary for effective binding.
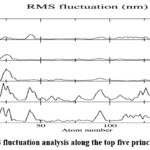 |
Figure 8: RMS fluctuation analysis along the top five principal componentsClick here to view Figure |
 |
Figure 9: 2D Projection of TrajectoryClick here to view Figure |
 |
Figure 10: Eigenvector component analysisClick here to view Figure |
The free energy calculations using both Poisson-Boltzmann (PB) and Generalized Born (GB) models provided critical insights into the stability and dynamics of the protein-ligand interaction. The PB analysis ( Fig 11) revealed a significant van der Waals contribution of -33.70 kcal/mol, indicative of strong hydrophobic interactions, and an electrostatic contribution of -100.65 kcal/mol, highlighting the importance of charged residue interactions in stabilizing the complex. Polar solvation energy was calculated as 98.75 kcal/mol, partially offsetting the binding energy due to desolvation penalties, while the nonpolar solvation contribution of -4.01 kcal/mol further stabilized the interaction. The total binding energy using the PB model was -39.61 kcal/mol, confirming robust ligand binding.
Similarly, the GB analysis ( Fig 12) yielded consistent results, with a van der Waals contribution of -33.70 kcal/mol and electrostatics of -100.65 kcal/mol. The polar solvation energy was slightly higher at 102.51 kcal/mol, while the nonpolar solvation contribution was -5.57 kcal/mol. The total free energy using the GB model was calculated as -37.41 kcal/mol, corroborating the PB results and reinforcing the strong binding affinity of Asiaticoside. Residue-specific energy decomposition further highlighted the critical roles of specific residues in ligand binding. Key residues such as LYS-114 (-18.56 kcal/mol) and LYS-121 (-17.01 kcal/mol) contributed significantly to stabilizing the protein-ligand complex through strong electrostatic interactions and hydrogen bonds. ASP-122, GLU-107, and ALA-118 also demonstrated notable contributions, reflecting their involvement in both hydrophobic and electrostatic stabilization.
Collectively, the free energy calculations demonstrate that the binding mechanism of Asiaticoside is driven predominantly by electrostatic interactions, with supplementary contributions from van der Waals forces and hydrophobic stabilization. These results confirm the strong and stable binding of Asiaticoside to the Cofilin dimer interface, supporting its potential as a potent inhibitor by disrupting critical interactions necessary for dimerization.
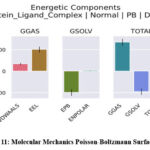 |
Figure 11: Molecular Mechanics Poisson-Boltzmann Surface AreaClick here to view Figure |
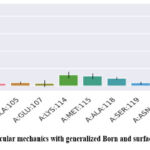 |
Figure 12: Molecular mechanics with generalized Born and surface area solvationClick here to view Figure |
Discussion
In this study, a cofilin dimerization inhibition process was explored as a tool for combating cognitive decline and neurodegenerative disorders.11 Under oxidative stress, cofilin dimers, formed through intermolecular disulfide bonds, switch cofilin’s primary function from actin filaments to promote bundling.12 In pathological contexts, such as neurodegenerative disorders where disrupted cytoskeletal dynamics lead to impaired cellular functions and acceleration of cognitive deterioration, this structural and functional transition makes a significant contribution.13 Asiaticoside natural compound derived from Bacopa monnieri (Brahmi)showed the greatest affinity (-7.3 kcal/mol) to the cofilin dimer interface. Other inhibitors with respect to actinorhodin (-6.9 kcal/mol) and granatin A (-6.8 kcal/mol) also stand as promising ones. These assembly studies with these compounds revealed strong dimerization disrupting interactions with residues critical for dimer stabilization, LYS-114, GLU-107, and ASN-138.
Molecular Dynamics and Mechanistic Insights The stability of these inhibitors was validated by molecular dynamics (MD) simulations, which had minimal RMSD fluctuations, robust hydrogen bonding, and consistent protein-ligand interactions during the simulation period. The mechanistic insight is consistent with a dichotomy between monomeric and dimeric states of cofilin. These inhibitors disrupt the dimerization interface and permit the preservation of cofilin in its active monomeric form, critical for severing actin filaments and maintaining cytoskeletal dynamics. Cofilin’s severing activity must be restored because dimeric cofilin promotes actin bundling, thereby impeding cellular motility and cytoskeletal remodeling. This approach demonstrates the pathological consequences of excessive actin bundling and therefore reduces the onset and the progression of cognitive decline in the cases of neurodegenerative disorders.
Free energy calculations showed that binding to Asiaticoside was dominated by electrostatic interactions, though with additional contribution by van der Waals forces and hydrophobic stabilization. At the residue level, energy decomposition revealed LYS-114, LYS-121, and ASP-122 to be critical for ligand binding and dimer disruption corroborated by MD simulations which showed stable conformational states and minor structural fluctuations of the protein-ligand complexes. A robust binding of Asiaticoside in cofilin was shown by showing its consistency in the hydrogen bond network, low RMSD, and SASA fluctuation, and thus its potential to effectively disrupt cofilin dimerization. A broader therapeutic implication of such a hit is provided by this study in targeting cofilin dimerization to prevent cognitive decline
. Neuronal plasticity and synaptic function that are essential for the maintenance of cognitive performance are impaired by excessive actin bundling driven by cofilin dimerization.14 The identified inhibitors prevent oxidative stress-induced cytoskeletal disruptions by preserving cofilin’s active state, thereby allowing neuroprotection. Asiaticoside identification from Brahmi is an instance of integrating traditions of medicinal knowledge with computational biology.15 The dual potential for cytoskeletal regulation and cognitive health represents a promising avenue of therapeutic interventions for oxidative stress and impaired cellular dynamics-based disorders. Comparisons with experimental data, and particularly exploration of how well these inhibitors alleviate oxidative stress under multiple conditions, are necessary to confirm and extend the computational findings. A deeper understanding of the impact of these on actin dynamics and neuroprotection will be gained by structural and functional assays in cellular systems. The translational potential of these compounds can be increased by optimizing the chemical properties of these compounds, particularly Asiaticoside, to improve bioavailability and target selectivity. This study demonstrates compelling proof of concept for targeting cofilin dimerization to prevent the onset and progression of cognitive decline. It develops a basis for innovative therapeutic strategies based on computational insights integrated with traditional medicinal knowledge, which can preserve cognitive health in neurodegenerative disorders.
Conclusion
Cofilin is critical for maintaining neuronal function, transitioning between monomeric and dimeric states to regulate actin filament behavior. This work combines computational insights with theoretical models of cofilin dynamics to highlight the therapeutic virtue of disrupting dimer formation. Natural products with cofilin inhibition activities were studied with the proposed possibility of delaying the onset of cognitive decline. Among the identified compounds, Asiaticoside—a natural product derived from Bacopa monnieri—emerged as the most prominent hit, exhibiting the strongest binding affinity (-7.3 kcal/mol), stable interaction with dimer interface residues, and robust structural integrity during simulation. These findings suggest that Asiaticoside could effectively disrupt cofilin dimerization and preserve the monomeric, actin-severing form, essential for maintaining cytoskeletal dynamics and synaptic function in neurons. Computational approaches including molecular docking and molecular dynamics simulations provided detailed insight into the dimer interface and molecular interactions that favor destabilization of the dimer complex. These results support a novel strategy in the design of small-molecule cofilin dimer inhibitors, contributing to therapeutic approaches that target early-stage mechanisms of cognitive decline. Future perspectives include experimental validation of these findings in vitro and in vivo, structural optimization of Asiaticoside to improve pharmacokinetic properties and exploration of its efficacy in oxidative stress-induced neurodegeneration models. Ultimately, this study provides a proof-of-concept for targeting cofilin dimerization as a viable neuroprotective strategy, integrating traditional medicinal compounds with modern computational drug discovery.
Acknowledgement
The author would like to thank Saveetha University for providing the necessary infrastructure and resources to carry out this research.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials
Permission to reproduce material from other sources
Not applicable
Author Contributions
- Magesh Mohan: Data Collection, Writing – Review & Editing.
- Renukadevi Jeyavelkumaran: Conceptualization, Methodology, Writing – Original Draft, Data Collection, Analysis
- Vijayakumar Balakrishnan; Writing – Review & Editing.
- Sanjay Valliappan: Methodology, Writing – Original Draft..
References
- Morozova A, Zorkina Y, Abramova O, et al. Neurobiological highlights of cognitive impairment in psychiatric disorders. Int J Mol Sci. 2022;23(3):1217. doi:10.3390/ijms23031217
CrossRef - Ojelade SA, Acevedo SF, Rothenfluh A. The role of the actin cytoskeleton in regulating Drosophila behavior. Rev Neurosci. 2013;24(5):471-484. doi:10.1515/revneuro-2013-0017
CrossRef - Lapeña-Luzón T, Rodríguez LR, Beltran-Beltran V, Benetó N, Pallardó FV, Gonzalez-Cabo P. Cofilin and neurodegeneration: New functions for an old but gold protein. Brain Sci. 2021;11(7):954. doi:10.3390/brainsci11070954
CrossRef - Bamburg JR, Minamide LS, Wiggan O, Tahtamouni LH, Kuhn TB. Cofilin and actin dynamics: Multiple modes of regulation and their impacts in neuronal development and degeneration. Cells. 2021;10(10):2726. doi:10.3390/cells10102726
CrossRef - Cichon J, Sun C, Chen B, et al. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J Biol Chem. 2012;287(6):3919-3929. doi:10.1074/jbc.M111.301911
CrossRef - Paciello F, Battistoni M, Martini S, et al. Role of LIMK1-cofilin-actin axis in dendritic spine dynamics in Alzheimer’s disease. Cell Death Dis. 2025;16(1):431. doi:10.1038/s41419-025-07741-7
CrossRef - Wurz AI, Schulz AM, O’Bryant CT, Sharp JF, Hughes RM. Cytoskeletal dysregulation and neurodegenerative disease: Formation, monitoring, and inhibition of cofilin-actin rods. Front Cell Neurosci. 2022;16:982074. doi:10.3389/fncel.2022.982074
CrossRef - Satish S. Molecular docking analysis of protein filamin-A with thioazo compounds. Bioinformation. 2023;19(1):99-104. doi:10.6026/97320630019099
CrossRef - Protim M, Afeeza K, Vasugi S, Dilipan E. Computational analysis of a marine-derived drug from Rhizophora mucronata against the capsid protein of rubella virus. Cureus. 2024;16(8):e67352. doi:10.7759/cureus.67352
CrossRef - Saini RS, Binduhayyim RIH, Gurumurthy V, et al. In silico assessment of biocompatibility and toxicity: molecular docking and dynamics simulation of PMMA-based dental materials for interim prosthetic restorations. J Mater Sci Mater Med. 2024;35(1). doi:10.1007/s10856-024-06799-7
CrossRef - Almarghalani DA, Bahader GA, Ali M, Tillekeratne LMV, Shah ZA. Cofilin inhibitor improves neurological and cognitive functions after intracerebral hemorrhage by suppressing endoplasmic reticulum stress related-neuroinflammation. Pharmaceuticals (Basel). 2024;17(1). doi:10.3390/ph17010114
CrossRef - Bamburg JR, Bernstein BW. Actin dynamics and cofilin-actin rods in alzheimer disease. Cytoskeleton (Hoboken). 2016;73(9):477-497. doi:10.1002/cm.21282
CrossRef - Ramasubburayan R, Amperayani KR, Varadhi G, et al. Unraveling bioactive metabolites of mangroves as putative inhibitors of SARS-CoV-2 Mpro and RBD proteins: molecular dynamics and ADMET analysis. J Biomol Struct Dyn. Published online October 28, 2023:1-10. doi:10.1080/07391102.2023.2275185
CrossRef - Borovac J, Bosch M, Okamoto K. Regulation of actin dynamics during structural plasticity of dendritic spines: Signaling messengers and actin-binding proteins. Mol Cell Neurosci. 2018;91:122-130. doi:10.1016/j.mcn.2018.07.001
CrossRef - Jeyasri R, Muthuramalingam P, Suba V, Ramesh M, Chen JT. Bacopa monnieri and their bioactive compounds inferred multi-target treatment strategy for neurological diseases: A cheminformatics and system pharmacology approach. Biomolecules. 2020;10(4):536. doi:10.3390/biom10040536
CrossRef
Abbreviations List
MM-PBSA – Molecular Mechanics Poisson-Boltzmann Surface Area,
MM-GBSA – Molecular Mechanics Generalized Born Surface Area,
RMSD – Root Mean Square Deviation

