
Manuscript accepted on :13-05-2025
Published online on: 29-05-2025
Plagiarism Check: Yes
Reviewed by: Dr. Kiruthika Balasubramanian
Second Review by: Dr. Swastika Maity
Final Approval by: Dr. Prabhishek Singh
Amna Makawi*1 , Somia Attaelseed Khalafallah2, Aymen Elfadil Abbas3, Asim Eisa4, Ahmed Mohammed Adam5, Israa Faris6 and Mohamed Alfaki 7
, Somia Attaelseed Khalafallah2, Aymen Elfadil Abbas3, Asim Eisa4, Ahmed Mohammed Adam5, Israa Faris6 and Mohamed Alfaki 7
1Department of Medicine, Elrazi University, Khartoum, Sudan.
2Department of Hematology and Immunohematology, Ibn Sina University, Khartoum, Sudan.
3Department of plastic surgery, Burjeel Medical City, Abu Dhabi, UAE.
4Department of Medicine, University of Bahri, Khartoum, Sudan.
5Department of biochemistry, Faculty of Science, King Abdulaziz University, Saudi Arabia.
6Department of Fertilization and Artificial Insemination, Istanbul University-Cerrahpaşa, Istanbul, Turkey.
7Department of Software Engineering, Al-Neelain University, Khartoum, Sudan.
Corresponding Author E-mail: makawiamna@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/3203
Abstract
Duchenne Muscular Dystrophy (DMD) is a severe genetic disorder characterized by progressive muscle degeneration, primarily affecting young boys and leading to significant disability and reduced life expectancy. Mutations in the DMD gene disrupt dystrophin production, triggering a cascade of molecular events that compromise muscle integrity. Despite advances in understanding DMD’s genetic basis, a comprehensive mapping of the molecular networks driving disease progression remains limited. This study addresses this gap through a bioinformatics analysis of gene expression datasets GSE38417 and GSE1004, sourced from the National Institutes of Health Gene Expression Omnibus (NCBI GEO) database. Using the GEO2R tool, we identified 135 differentially expressed genes (DEGs) associated with DMD, revealing distinct patterns of upregulation and downregulation that reflect the disease’s complex pathophysiology. To investigate the interactions among these DEGs, we constructed protein-protein interaction (PPI) networks utilizing the STRING database and Cytoscape software, enabling the identification of 10 hub genes, including SPP1 and POSTN, central to DMD’s molecular architecture. Enrichment analysis, conducted via the Reactome database, associated these hub genes with the extracellular matrix organization pathway, highlighting its essential role in maintaining muscle structure and its dysregulation in DMD. To ensure the robustness of our findings, we validated the DEGs by cross-referencing with data from OMIM and GeneCards, confirming the involvement of these genes and the extracellular matrix organization pathway in DMD pathology. Additional hub genes (e.g., SGCA, SGCD) from OMIM and GeneCards highlighted the importance sarcolemma stability. This study elucidates critical molecular drivers of DMD and underscores potential therapeutic targets, laying a groundwork for future research aimed at mitigating disease progression and enhancing patient outcomes through targeted interventions.
Keywords
Bioinformatics; Duchenne muscular dystrophy; Dystrophin protein; Enrichment pathways; hub genes; Extracellular matrix organization
Download this article as:| Copy the following to cite this article: Makawi A, Khalafallah S. A, Abbas E. A, Eisa A, Adam A. M, Adam I, Alfaki M, Molecular Architecture of Duchenne Muscular Dystrophy: Hub Gene Identification and Functional Pathway Analysis. Biomed Pharmacol J 2025;18(2). |
| Copy the following to cite this URL: Makawi A, Khalafallah S. A, Abbas E. A, Eisa A, Adam A. M, Adam I, Alfaki M, Molecular Architecture of Duchenne Muscular Dystrophy: Hub Gene Identification and Functional Pathway Analysis. Biomed Pharmacol J 2025;18(2). Available from: https://bit.ly/44TYXd6 |
Introduction
The most prevalent and severe kind of muscular dystrophy is Duchenne muscular dystrophy (DMD), which causes mobility issues, the eventual requirement for assisted ventilation, and early mortality. 1 According to a recent comprehensive review, the global birth prevalence was 19.8 per 100,000 live male births, while the global DMD prevalence was 7.1 cases per 100,000 boys. It is brought on by mutations that interfere with the dystrophin protein’s synthesis .2
Situated on chromosome Xp21.1, the dystrophin gene is one of the biggest genes in the human body. It spans 2.2 million base pairs and has seven promoters and 79 exons.3,4 More than 1,100 mutations have been found so far, 891 of which are the cause of DMD symptoms.5 While some alterations are secondary to point mutations or frameshift rearrangements, the vast majority (60%) of these mutations are caused by massive insertions or deletions of the gene.6 The rod-shaped cytoplasmic protein dystrophin acts as a link between the dystroglycan complex and the cell’s extracellular matrix and intracellular contractile apparatus. 7
Global population-based research has shown that the average age of diagnosis is around 5 years old. 8 A helpful, but underutilized, screening test for people suspected of having DMD is the creatine kinase (CK) level, which is nearly invariably increased in DMD patients. 9 In the first phase of the disease, the child experiences difficulty in running, climbing stairs, jumping, getting up from the ground, falls frequently, and develops a waddling gait with a positive “Gowers’ sign”. 10 On average, untreated patients lose their ability to walk independently between 8 and 12 years, and with the progression of the disease scoliosis and breathing difficulties occur. 11 Cardiac and orthopaedic complications are common, and death usually occurs in the twenties due to respiratory muscle weakness or cardiomyopathy. 12,13 However, the advent of corticosteroids, mechanical breathing, cardiac management, spine surgery, and multidisciplinary treatment has extended the life expectancy of DMD patients in recent decades. 14,15,16
Early diagnosis can open up access to cutting-edge therapies and clinical trials, as well as empower parents to make well-informed family planning decisions. 17 In the future, sensitive biomarkers, natural history data, and improved trial designs may help overcome the challenges of clinical development brought on by the rarity and unpredictability of Duchenne muscular dystrophy. 18
Although there is currently no cure for DMD patients’ declining muscle function, treatment options include scoliosis correction surgery, assisted cough and ventilation support, ankle-foot orthoses, cardiac management, and psychosocial care, all of which can enhance quality of life. 19 Gene transfer to restore dystrophin expression using a safe, non-pathogenic viral vector known as an adeno-associated viral (AAV) vector is a viable treatment strategy for this deadly illness. 20 In this study, bioinformatics methods were used to identify potential hub genes related to DMD. Despite advances in DMD research, the integration of diverse datasets to comprehensively map hub genes and pathways remains limited. This study addresses this gap by using bioinformatics methods and combining these datasets to uncover novel molecular insights into DMD pathogenesis. We hope to provide insights that improve treatment strategies and help patients achieve better outcomes.
Materials and Methods
Dataset selection and differentially expressed gene identification
This study utilized two datasets, GSE38417 and GSE1004, obtained from the National Institutes of Health Gene Expression Omnibus (NCBI GEO) database (https://www.ncbi.nlm.nih.gov/geo/), a publicly accessible repository for gene expression data. 21
These datasets were chosen to investigate differentially expressed genes (DEGs) associated with DMD. DEGs between samples and controls were identified via the GEO2R tool (https://www.ncbi.nlm.nih.gov/geo/geo2r/), an interactive web application that leverages the limma package in R and GEO query for differential expression analysis. The data were normalized with Robust Multi-array Average (RMA), a method suited for Affymetrix platforms. To ensure consistency and minimize batch effects, datasets were processed uniformly, although GEO2R does not inherently correct for such variations. We set a |log2FC| > 1.5 threshold criteria, which is a widely accepted standard in gene expression studies to spot important gene changes, and used an adjusted p-value < 0.01 with the Benjamini-Hochberg method for reliability. To validate these DEGs, we cross-referenced them with known DMD-related genes from the Online Mendelian Inheritance in Man (OMIM) database (https://www.omim.org/) and the GeneCards database (https://www.genecards.org/). Overlapping genes were identified to confirm their relevance to DMD pathology.
Protein‒protein interaction network construction and identification of hub genes.
To analyze protein‒protein interactions (PPIs), the STRING database (version 12.0) (https://string-db.org/) was used. STRING integrates known and predicted protein‒protein association data, facilitating comprehensive assessments of these interactions. 22 The constructed PPI network was then imported into Cytoscape (version 3.10.2) for further analysis. Within Cytoscape, the degree algorithm was applied to identify hub genes and critical nodes with numerous connections in the network.
Hub genes and cluster identification
Hub genes within the PPI network were identified via the cytoHubba plugin in Cytoscape (https://cytoscape.org/). 23 This plugin ranks genes based on their connectivity and significance within the network. The degree algorithm filtered and ranked these hub genes, highlighting those with the most significant interactions relevant to DMD.
Enrichment analysis
The Reactome database (https://reactome.org/) was utilized to explore the biological functions and signaling pathways of the identified hub genes. The Reactome is a comprehensive, open-source resource that provides detailed information on various biological pathways. 24 It supports the visualization, interpretation, and analysis of pathway data, aiding research in areas such as signal transduction, immune system function, and metabolism. Additionally, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (https://www.genome.jp/kegg/) for enrichment analysis. Gene Ontology (GO) analysis, covering molecular function, cellular component, and biological process domains, was conducted using the GO Consortium’s enrichment tool (http://geneontology.org/). Relationships between DEGs, hub genes, and enriched pathways were visualized using Sanky plots.
Results
Expression of differentially expressed genes from NCBI GEO datasets
We used two GSE datasets, GSE38417 and GSE1004. According to the GSE38417 dataset, 22,185 genes were set at |log2FC| > 1.5 and adjusted p value < 0.01. We identified 1715 significant genes, of which 417 were downregulated and 1,298 genes were upregulated. The analysis of the GSE1004 dataset yielded 9153 genes that were within the threshold of |log2FC| > 1.5 and an adjusted p-value < 0.01. These results indicated that among the genes identified, there was a trend of upregulation in 1298 genes and a trend of downregulation in 167 genes. Using the R platform, we illustrated the regulation of genes for both the NCBI s, namely, GSE38417 and GSE1004, as shown in Figures 1A and 1B. We then plotted the datasets in a Venn diagram, as shown in Figure 1C, to establish the genes common to the two datasets, and we found that there were 135 common genes. A total of 120 genes were upregulated, and 11 genes were downregulated, showing widespread transcriptional dysregulation in DMD. The pattern of regulation was the same for the 135 genes except for four genes namely PRMT2, MYL4, C14orf132, and BICD1, which had different regulation patterns between the two databases. We chose two public datasets, namely, OMIM and GeneCards, to further understand the molecular and genetic mechanisms of the disease. The DEGs between the two datasets were identified and plotted in a Venn diagram (Figure 1D). We filtered out 226 genes in the OMIM database and 782 genes from GeneCards; 62 were differentially expressed genes, highlighting key genetic factors involved in DMD development.
 |
Figure 1: Shows the differentially expressed genes located in the NCBI GEO database and public datasets alongside OMIM and GeneCards.Click here to view Figure |
Gene Ontology and functional enrichment analysis of differentially expressed genes
The three core domains of Gene Ontology include the molecular function, cellular component, and biological processes. As shown in Figure 2A, we studied the gene ontologies of the filtered genes from the NCBI GEO datasets GSE38417 and GSE1004. The most common molecular function was protein kinase activator activity, but this effect was not significant (log2FC < 1.5, p > 0.01). In terms of the cellular component, the collagen-containing extracellular matrix was the most common component present in most of the genes, and it was found to be significant (log2FC > 1.5, p < 0.01), as indicated by the red dots in Figure 2A. Extracellular matrix organization was the most common biological process indicated by the green dots, which is expected and central result of our analysis. Similarly, Figure 2B highlights the gene ontology of the differentially expressed genes from OMIM and the GeneCards. Sodium channel regulator activity is the most common molecular function; however, this function was not found to be significant (log2FC > 1.5, p < 0.01). The most common cellular component was the sarcolemma, which was significant (log2FC > 1.5, p < 0.01). Muscle organ development was the most common biological process according to the DEGs from OMIM and the GeneCards (log2FC > 1.5, p < 0.01). Additionally, we presented the differentially expressed genes in a Sanky plot to understand the functional enrichment analysis. From Figure 2C and 2D, we can conclude that even though the differentially expressed genes from NCBI geo and OMIM and the GeneCards were different, they shared the same enrichment pathway, which was extracellular matrix organization and was found to be significant (log2FC > 1.5, p < 0.01).
 |
Figure 2: Gene ontology and enrichment pathway analysis of the differentially expressed genes.Click here to view Figure |
A: Gene ontology of differentially expressed genes from the NCBI GEO database. B: Gene ontology of differentially expressed genes from OMIM and the gene card. C: Sanky plot highlighting the enrichment pathways of differentially expressed genes from the NCBI Geo database. D: Sanky plot highlighting the enrichment pathways of differentially expressed genes from OMIM and the GeneCards.
Count: number of genes involved in the pathway; red dots: more significant (p < 0.01); green dots: less significant (p > 0.01)
Protein‒protein interaction and hub gene identification:
Here, we constructed a protein‒protein interaction (PPI) network of overlapping differentially expressed genes via the STRING database, as shown in Figure 3. The PPI constructed from the NCBI GEO database had 129 nodes and 386 edges, identified 10 hub genes, including SPP1 and POSTN, suggesting a central role in ECM organization (Figure 3C). The average node degree was 5.98, and the PPI enrichment P value was less than 1.0e-16. Similarly, we constructed a PPI from the overlapping differentially expressed genes from OMIM and the GeneCards, which had 58 nodes and 290 edges, denoted 10 hub genes, such as SGCA and SGCD, linked to sarcolemma stability (Figure 3D). The average node degree was 10, and the PPI enrichment P value was less than 1.0e-16.
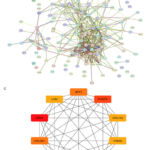 |
Figure 3: Protein‒protein interaction and hub gene identification via a STRING network and Cytoscape.Click here to view Figure |
A: Protein‒protein interactions of the differentially expressed genes from the NCBI GEO database. B: Protein‒protein interactions of the DEGs from OMIM and the GeneCards. C: Hub genes identified from the DEGs obtained from NCBI GEO database D: Hub genes identified from the DEGs obtained from OMIM and the GeneCards.
Functional enrichment analysis and gene ontology of hub genes
Gene Ontology encompasses three main domains, which are the molecular function, cellular component, and biological processes. As demonstrated in Figure 4A, we analyzed the gene ontologies related to the hub genes from the NCBI GEO datasets. The most common molecular function identified was protease binding, although it had low significance (log2FC < 1.5, p > 0.01). With respect to the cellular component, the collagen-containing extracellular matrix was the most prevalent among the hub genes and was significant (log2FC > 1.5, p < 0.01), as indicated by the red dots in Figure 4A. The most common biological process was extracellular matrix organization, which is marked by a green dot.
Similarly, figure 4B illustrates the gene ontologies of the hub genes from OMIM and GeneCards. Vinculin binding was the most common molecular function, but it was not significant (log2FC, p > 0.01). The sarcolemma was the most common cellular component and was significant (log2FC > 1.5, p < 0.01). Muscle organ development was the most common biological process associated with the expressed hub genes from OMIM and GeneCards (log2FC > 1.5, p < 0.01).
Additionally, we examined the enriched pathways via the Reactome database. Figure 4C shows the enrichment pathways related to the hub genes from the NCBI GEO datasets, with the most common being extracellular matrix organization (R-HSA-1474244), which was significant (p < 0.01). As shown in Figure 4D, we found that EGR2- and SOX10-mediated initiation of Schwann cell myelination (R-HSA-9619665) is a significant pathway for hub genes from OMIM and GeneCards (p < 0.01).
 |
Figure 4: Gene Ontology and enrichment pathway analysis of the hub genesClick here to view Figure |
A: Gene Ontology of the hub genes from the NCBI GEO database. B: Gene Ontology of the hub genes from OMIM and the GeneCards. C: Bar chart highlighting the enrichment pathways of hub genes from the NCBI GEO database. D: Bar chart showcasing the enrichment pathways of the hub genes from OMIM and the GeneCards.
Discussion
This study identified 135 DEGs shared between the NCBI GEO datasets GSE38417 and GSE1004 and 62 overlapping DEGs between the OMIM and Gene Cards datasets, the majority of which were upregulated, which emphasizes a strong and consistent molecular pattern associated with DMD. Gene Ontology enrichment revealed significant biological processes, including organization of the ECM and muscle organ development, whereas collagen-rich ECM and sarcolemma comprised major cellular components. By analyzing PPIs, significant connectivity was obtained among the DEGs, which indicated that these DEGs are involved in the development of the disease. The results reflect the molecular mechanisms driving DMD and suggest some potential therapeutic targets for further investigations.
The results from previous studies are closely aligned with our findings, particularly regarding the organization of the ECM and the identification of important hub genes. 25, 26 One of these studies used the datasets GSE465, GSE1004, and GSE1007 to identify focal adhesion, PI3K-Akt signaling, calcium signaling, and ECM-receptor interaction as important pathways involved in the development of DMD. This has revealed the relationships among the degradation of the ECM, cellular signaling pathways, and the integrity of muscle fibers. Our study did not identify these pathways; however, this may be explained by the different datasets used or methodological approaches employed, which suggests that ECM organization may dominate in our cohorts, offering a distinct molecular perspective on DMD progression. However, these findings align with the broad concept of signaling pathways and extracellular matrix dysfunction. Several important hub genes, including THBS2, COL1A2, and COL3A1, which support our findings, have been recognized as crucial regulators of ECM structure and fibrosis. 25 These genes are associated with excessive accumulation of collagen, one of the hallmark features of severe muscle disorders such as DMD. They are vital to the integrity and proper function of muscles. Our research extends on prior results by analyzing a wider variety of datasets and identifying novel biological processes that are critical for the progression of DMD, such as the sarcolemma and muscle organ development.
Recent comprehensive reviews further illustrate the role of extracellular matrix disruption and calcium signaling in DMD etiology. 27, 28 These studies provide detailed molecular explanations of how a deficiency in dystrophin leads to abnormal calcium handling. It disrupts the connections between the cytoskeleton and the extracellular matrix, causing progressive muscle degeneration.
Although these results highlight the pathophysiological basis of DMD, the development of the illness also significantly involves immunological dysregulation. The following study, 29 illuminates how immunological systems work in DMD. This study identified hub genes highly elevated in DMD patients via weighted gene expression network analysis (WGCNA) that were enriched in immune-related pathways such as complement activation and immune effector regulation. These genes included SERPING1, F13A1, C1S, C1R, and HLA-DPA1. These findings are of interest because they highlight the crosstalk between immune dysregulation and ECM remodeling in promoting disease progression,29 which complements our focus on extracellular matrix organization.
In addition to structural and immune-related factors, regulatory networks provide more understanding of the pathogenesis of DMD. A previous study,30 provided a different perspective by identifying DEGs and constructing a coregulatory network that included miRNAs, transcription factors, and DEGs. These findings further highlight the role of microRNAs, such as miR-124a and miR-200b/200c/429, and transcription factors, such as ZNF362 and SPI1, in controlling the pathways related to DMD.
Three genes are recognized: HERC5, SKP2, and FBXW5, which take part in mechanisms contributing to myofibril degeneration and muscle weakness, including protein polyubiquitination and ubiquitin-dependent protein degradation. 30 These findings complement our study by revealing a link between ECM organization and upstream regulatory networks involving microRNAs and transcription factors. Taken together, these findings reveal the interaction between DEG and epigenetic regulators in the context of DMD.
Therapeutic implication of key hub genes.
DMD is caused mainly by mutations in the dystrophin gene, leading to breakdown in muscle architecture and causing progressive loss of function. New strategies targeting specific mutations, including gene transfer and exon-skipping techniques, have been developed with the hope of correcting such mutations and improving patient outcomes. However, effective delivery of these therapies into muscle tissue represents a major challenge. 31
Novel approaches, such as systemic delivery of stem cells for targeted repair and gene correction for specific mutations, hold considerable therapeutic promise; however, further optimization is necessary because of the heterogeneity of the mutations responsible for DMD. 32 These obstacles call for complementary strategies that target broader pathological mechanisms, including ECM remodeling, immune dysregulation, and sarcolemma instability. Our analysis revealed a complex network of molecular interactions other than primary genetic defects, revealing key hub genes involved in the disease course and avenues for therapeutic interventions.
Among the most promising therapeutic targets in DMD, periostin is considered a key player in the process of fibrosis and remodeling of the ECM. Increased levels of periostin, especially in younger patients, are known to contribute to abnormal ECM deposition and muscle degeneration. The removal of periostin in mouse models has been shown to decrease fibrosis and improve muscle regeneration by modulating the TGF-β pathway, which is critical for the progression of DMD. This dual effect, i.e., less fibrosis combined with enhanced tissue repair indicates that periostin holds promise as a target for novel therapies aimed at lowering fibrosis without impairing ECM stability or regeneration. 33, 34
The macrophage migration inhibitory factor (MIF) pathway is a favorable target for DMD therapy because of its involvement in regulating inflammation and ECM remodeling. MIF and its receptors, namely, CD74, CD44, and CXCR4, are significantly elevated in DMD, activating proinflammatory pathways such as the ERK1/2 MAP kinase, cPLA2, and NF-κB pathways, which lead to persistent inflammation and fibrosis. Gene expression analysis revealed that MIF-related genes such as CD44 promote the development of disease through ECM organization and fibrosis; all these findings point to early onset disease mechanisms. 35 MIF has been targeted with small molecule inhibitors, monoclonal antibodies, and peptide inhibitors and has shown promise in reducing inflammation and fibrosis in preliminary studies. In addition, the modulation of downstream pathways, such as the NF-κB pathway, may increase the efficacy of treatment. However, there are still some obstacles in optimizing the drug delivery system and reducing undesirable side effects, which hinders its clinical application. 35, 36
The dystrophin-glycoprotein complex (DGC) can maintain muscle fiber structural integrity and transmit mechanical forces accurately to its sarcoglycan constituents. 37 Genetic mutations affecting the specific sarcoglycan genes SGCA and SGCD cause distinct forms of limb-girdle muscular dystrophies, LGMD2D and LGMD2F, respectively. 37, 38 Both conditions share major pathological features that are also characteristic of DMD, including compromised membrane stability, progressive fibrosis, and muscle degeneration. 38 Other emerging therapeutics have been demonstrated recently. One approach utilizes siRNAs targeting ECM-associated genes such as COL1A2, COL3A1, and FN1 to decrease the extent of fibrosis, which is common in DMD and is also observed in sarcoglycanopathies. Lipid nanoparticles (LNPs), particularly selective organ-targeting (SORT) nanoparticles, have been suggested to function effectively as siRNA delivery vehicles. By modifying the molecular composition of these nanoparticles so that they bind specifically to certain serum proteins, siRNAs can be delivered to muscle tissues—a positive approach to reduce fibrosis and restore muscle function. 26
Another approach involves gene replacement, specifically the restoration of functional SGCA via viral vector delivery. 39 This method has demonstrated long-term protein expression in clinical studies 40 and has shown long-term muscle protection in animal studies. 41 Another line of research has focused on Caveolin-3 (Cav3), a muscle-specific protein that modulates dystroglycan metabolism as well as the function of the sarcolemma. Observations from DMD patients and mdx mouse models have revealed altered Cav3 expression patterns, indicating its potential as a therapeutic target aimed at stabilizing the sarcolemma. 42
Utrophin (UTRN) is considered a very promising therapeutic target for DMD. Utrophin is the structural homolog of dystrophin, and its upregulation is a natural adaptive response in DMD since elevated expression of utrophin is associated with milder disease phenotypes. Thus, it could represent a therapeutic target suitable for all DMD patients, irrespective of mutation type. Current methods to increase utrophin include adeno-associated virus (AAV)-mediated gene delivery, small molecule up regulators, and transcriptional modulators, which have demonstrated preclinical effectiveness in enhancing muscle function in DMD. 43 These results suggest that a successful therapeutic strategy should integrate dystrophin-targeted treatments with methods that address multiple pathological processes. We highlight the necessity of multi-targeting strategies that consider tissue-specific impacts and personalized medicine tailored to individual patient genetics and disease progression.
Implications of the study
The shared pathway of ECM organization has become a promising therapeutic target for diseases driven by ECM dysregulation, including conditions such as fibrosis, cancer, and chronic inflammatory diseases. In addition, ECM components, including collagen, play pivotal roles in cellular signaling and tissue repair; this pathway may serve as a good candidate for therapeutic intervention. The identification of key hub genes, including POSTN, MIF, SGCA, SGCD, CAV3, and UTRN, highlights the most important mechanisms of DMD progression.
The pathogenesis of DMD involves ECM remodeling and sarcolemma integrity. These pathways can be targeted by inhibitors of periostin and MIF or modulators of utrophin, which have considerable therapeutic effects. For example, deficiency of periostin causes a reduction in fibrosis and enhances muscle regeneration by modifying TGF-β signaling. Similarly, DGC can also be stabilized by targeting sarcoglycan components such as SGCA and SGCD or caveolin-3 at CAV3 as a complementary approach for mitigating muscle damage.
Moreover, immune dysregulation facilitates DMD progression through mechanisms involving complement activation, and chronic inflammation plays an important role. Such a strategy, which integrates approaches to both ECM remodeling and immune signaling, could offer a comprehensive approach for the management of this disease.
Limitations of the Study
The study provided information about the molecular basis of DMD, although not without several limitations, including reliance on publicly available datasets that might represent potential variability owing to differences in sample characteristics and platform biases. Second, the present analysis is in-silico; hence, experimental validation is needed to confirm the identified hub genes for their possible role as therapeutic targets. The use of qRT‒PCR and Western blot analysis in future studies can confirm the expression of the hub genes. These findings can also be applied in functional studies of DMD models to further understand the roles of these genes in disease mechanisms. Advanced gene-editing techniques such as CRISPR-Cas9 and RNA therapeutics can theoretically provide specific treatments that target SGCA, SGCD, or UTRN.
Conclusion
In this study, we identified 10 important hub genes and related pathways associated with the pathogenesis of Duchenne muscular dystrophy. These findings emphasize that the organization of the extracellular matrix pathway and the disruption of sarcolemma integrity are key mechanisms promoting the disease process. Potential therapeutic targets include, but are not limited to, the central genes POSTN, MIF, SGCA, SGCD, CAV3, and UTRN. Targeting these genes might improve patient outcomes. This study provides the foundation for personalized medicine that correlates the genetics and disease stages of individual patients and provides a molecular framework for the progression of DMD.
Acknowledgement
I would like to thank all my mentors from the bioinformatics internship course who have supported me during the process of the manuscript writing.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials
Permission to reproduce material from other sources
Not Applicable
Author Contributions
- Amna Makawi: Initial manuscript blueprint, abstract, result extraction and database selection, complete paper review.
- Somia Attaelseed Khalafallah: Result extraction
- Aymen Elfadil Abbas: Introduction and discussion, paper review and modification
- Asim Eisa: Discussion, paper review and modification
- Ahmed Mohammed Adam: materials and methods, results writing contribution, paper review.
- Israa Faris: Reference, figure ligands
- Mohamed Alfaki: Supervisor of the paper, complete paper review and feedback before submission process.
References
- Lorentzos, M., Parsons, J. A., Jones, K. J., Servais, L., & Treat NMD Early Diagnosis Workshop Participants. Early diagnosis of Duchenne muscular dystrophy – A Treat-NMD international workshop. 2024, Neuromuscular disorders: NMD; 45: 104467.
CrossRef - Orso, M., Migliore, A., Polistena, B., et al. Duchenne muscular dystrophy in Italy: A systematic review of epidemiology, quality of life, treatment adherence, and economic impact. PloS one. 2023; 18(6): e0287774.
CrossRef - Geng, C., Zhang, C., Li, P., Tong, et al. Identification and characterization of two DMD pedigrees with large inversion mutations based on a long-read sequencing pipeline. European journal of human genetics: EJHG. 2023; 31(5): 504–511.
CrossRef - Wootton, M. Expansion of the replicative lifespan of human muscle cells by retroviral transduction of the catalytic telomerase subunit gene (Doctoral dissertation, University of Glasgow). 2004
- Capelletti, S., García Soto, S. C., & Gonçalves, M. A. F. V. On RNA-programmable gene modulation as a versatile set of principles targeting muscular dystrophies. Molecular therapy: the journal of the American Society of Gene Therapy. 2024; 32(11): 3793–3807.
CrossRef - Maki H. Origins of spontaneous mutations: specificity and directionality of base-substitution, frameshift, and sequence-substitution mutagenesis. Annual review of genetics. 2002; 36: 279–303.
CrossRef - Dowling, P., Gargan, S., Murphy, S., et al. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes. 2021; 9(1): 9.
CrossRef - Strømme, P., & Diseth, T. H. Prevalence of psychiatric diagnoses in children with mental retardation: data from a population-based study. Developmental medicine and child neurology. 2000; 42(4): 266–270.
CrossRef - Thomas, S., Conway, K. M., Fapo, O., et al. Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). Time to diagnosis of Duchenne muscular dystrophy remains unchanged: Findings from the Muscular Dystrophy Surveillance, Tracking, and Research Network, 2000-2015. Muscle & nerve. 2022; 66(2): 193–197.
CrossRef - Rideau, Y., Duport, G., Delaubier, A., et al. Early treatment to preserve quality of locomotion for children with Duchenne muscular dystrophy. Seminars in neurology. 1995; 15(1): 9–17.
CrossRef - Mohar, J. Untreated Early Onset Scoliosis-The Natural Progression of a Debilitating and Ultimately Deadly Disease. Recent Advances in Scoliosis. London: IntechOpen. 2012; 311-328.
CrossRef - Birnkrant, D. J., Bushby, K., Bann, C. M., et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. The Lancet Neurology. 2018; 17(4): 347-361.
CrossRef - Finder, J. D. Respiratory complications in neuromuscular disorders. Neuromuscular Disorders, 2022; 40-51.
CrossRef - Ryder, S., Leadley, R. M., Armstrong, N., et al.The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet journal of rare diseases. 2017; 12(1): 79.
CrossRef - Goemans, N., & Buyse, G. Current treatment and management of dystrophinopathies. Current treatment options in neurology. 2014; 16(5): 287.
CrossRef - Bushby, K., Bourke, J., Bullock, R., et al.. The multidisciplinary management of duchenne muscular dystrophy. Current Paediatrics. 2005; 15(4): 292-300.
CrossRef - Birnkrant, D. J., Bushby, K., Bann, C. M., et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. The Lancet. Neurology. 2018; 17(5): 445–455.
CrossRef - Liu, J., Barrett, J. S., Leonardi, E. T., et al. Natural History and Real-World Data in Rare Diseases: Applications, Limitations, and Future Perspectives. Journal of clinical pharmacology. 2022; 62 Suppl 2(Suppl 2): S38–S55.
CrossRef - D’Amario, D., Amodeo, A., Adorisio, R., et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart (British Cardiac Society). 2017; 103(22): 1770–1779.
CrossRef - Elangkovan, N., & Dickson, G. Gene Therapy for Duchenne Muscular Dystrophy. Journal of neuromuscular diseases. 2021; 8(s2): S303–S316.
CrossRef - Barrett, T., Wilhite, S. E., Ledoux, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Research. 2012; 41(D1): D991–D995.
CrossRef - Szklarczyk, D., Kirsch, R., Koutrouli, M., et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic acids research. 2023; 51(D1): D638–D646.
CrossRef - Shannon, P., Markiel, A., Ozier, O., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003; 13(11): 2498–2504.
CrossRef - Croft, D., O’Kelly, G., Wu, G., et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Research. 2010; 39(Database): D691-D697.
CrossRef - Zhang, R., Lv, L., Ban, W., Dang, X., & Zhang, C. Identification of Hub Genes in Duchenne Muscular Dystrophy: Evidence from Bioinformatic Analysis. Journal of computational biology: a journal of computational molecular cell biology. 2020; 27(1): 1–8.
CrossRef - Li, N., Xiahou, Z., Li, Z., Zhang, Z., Song, Y., & Wang, Y. Identification of hub genes and therapeutic siRNAs to develop novel adjunctive therapy for Duchenne muscular dystrophy. BMC Musculoskeletal Disorders. 2024; 25(1).
CrossRef - Mareedu, S., Million, E. D., Duan, D., & Babu, G. J. Abnormal calcium handling in Duchenne Muscular dystrophy: Mechanisms and potential therapies. Frontiers in Physiology. 2021; 12.
CrossRef - Zabłocka, B., Górecki, D. C., & Zabłocki, K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. International journal of molecular sciences. 2021; 22(20): 11040.
CrossRef - Wei, Y., Su, Q., & Li, X. Identification of hub genes related to Duchenne muscular dystrophy by weighted gene co-expression network analysis. Medicine. 2022; 101(52): e32603.
CrossRef - Xiu, M. X., Zeng, B., & Kuang, B. H. Identification of hub genes, miRNAs and regulatory factors relevant for Duchenne muscular dystrophy by bioinformatics analysis. The International journal of neuroscience. 2022; 132(3): 296–305.
CrossRef - Patterson, G., Conner, H., Groneman, M., et al. Duchenne muscular dystrophy: Current treatment and emerging exon skipping and gene therapy approach. European journal of pharmacology. 2023; 947: 175675.
CrossRef - Nowak, K. J., & Davies, K. E. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO reports. 2004; 5(9): 872–876.
CrossRef - Lorts, A., Schwanekamp, J. A., Baudino, T. A., McNally, E. M., & Molkentin, J. D. Deletion of periostin reduces muscular dystrophy and fibrosis in mice by modulating the transforming growth factor-β pathway. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109(27): 10978–10983.
CrossRef - Trundle, J., Cernisova, V., Boulinguiez, A., et al. Expression of the Pro-Fibrotic Marker Periostin in a Mouse Model of Duchenne Muscular Dystrophy. Biomedicines. 2024; 12(1): 216.
CrossRef - Lombardo, S. D., Mazzon, E., Mangano, K., et al. Transcriptomic Analysis Reveals Involvement of the Macrophage Migration Inhibitory Factor Gene Network in Duchenne Muscular Dystrophy. Genes. 2019; 10(11): 939.
CrossRef - Xin, J., & Liu, S. Identifying hub genes and dysregulated pathways in Duchenne muscular dystrophy. The International journal of neuroscience, 1–13. Advance online publication. 2024.
CrossRef - Gonzalez-Quereda, L., Gallardo, E., Töpf, A., et al. A new mutation of the SCGA gene is the cause of a late onset mild phenotype limb girdle muscular dystrophy type 2D with axial involvement. Neuromuscular disorders: NMD. 2018; 28(8): 633–638.
CrossRef - Reddy, H. M., Hamed, S. A., Lek, M., et al. Homozygous nonsense mutation in SGCA is a common cause of limb-girdle muscular dystrophy in Assiut, Egypt. Muscle & nerve. 2016; 54(4): 690–695.
CrossRef - Mendell, J. R., Rodino-Klapac, L. R., Rosales-Quintero, X., et al. Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins. Annals of neurology. 2009; 66(3): 290–297.
CrossRef - Mendell, J. R., Rodino-Klapac, L. R., Rosales, X. Q., et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Annals of neurology. 2010; 68(5): 629–638.
CrossRef - Pacak, C. A., Walter, G. A., Gaidosh, G., et al. Long-term skeletal muscle protection after gene transfer in a mouse model of LGMD-2D. Molecular therapy: the journal of the American Society of Gene Therapy. 2007; 15(10): 1775–1781.
CrossRef - Pradhan, B. S., & Prószyński, T. J. (2020). A Role for Caveolin-3 in the Pathogenesis of Muscular Dystrophies. International journal of molecular sciences. 2020; 21(22): 8736.
CrossRef - Soblechero-Martín, P., López-Martínez, A., de la Puente-Ovejero, L., Vallejo-Illarramendi, A., & Arechavala-Gomeza, V. Utrophin modulator drugs as potential therapies for Duchenne and Becker muscular dystrophies. Neuropathology and applied neurobiology. 2021; 47(6): 711–723.
CrossRef

