
Manuscript accepted on :19-05-2025
Published online on: 05-06-2025
Plagiarism Check: Yes
Reviewed by: Dr. Arif Ansori
Second Review by: Dr. Avinash Kumar Singh
Final Approval by: Dr. Patorn Piromchai
Hasanain Abdulhameed Odhar* , Azher Abdulmutaleb Ibrahim, Ahmed Fadhil Hashim
, Azher Abdulmutaleb Ibrahim, Ahmed Fadhil Hashim
Department of Pharmacy, Al-Zahrawi University College, Karbala, Iraq
Corresponding Author E-mail: hodhar3@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/3196
Abstract
The influenza virus is a highly mutagenic pathogen that can drive a pandemic threat. This pandemic threat is currently very probable due to the recent outbreak of avian influenza H5N1 among cattle herds in United States. It is possible that this pathogenic strain can undergo evolution to easily infect other mammals including human beings. Therefore, it is of interest to identify novel antiviral molecules that can fight both the circulating or novel strains of influenza. In this in-silico study, a library of Traditional Chinese Medicine (TCM) was screened against neuraminidase enzyme for H5N1 influenza virus. Both molecular docking and dynamics simulation were used to identify potential inhibitors for N1 neuraminidase protein. According to the results of this study, both grosvenorine and pectolinarin are predicted to be effective N1 neuraminidase inhibitors. Evaluation of docking images points to the possibility that these two flavonoids may be engaged in multiple interactions with key residues in the active site of neuraminidase monomer. Additionally, the two compounds were able to record close proximity to target active site during simulation study. Also, the minimal average molecular mechanics-Poisson Boltzmann surface area (MM-PBSA) binding energy was reported by pectolinarin throughout simulation interval. Finally, chemical and pharmacokinetics profiling refers to the possible hydrophilic nature of both grosvenorine and pectolinarin. Thus, these two compounds are supposed to be soluble in water with low intestinal absorption. Also, both compounds are anticipated to be safe with high median lethal dose (LD50). However, these preliminary computation results must be further evaluated both in-vitro and in-vivo.
Keywords
Docking; Dynamics simulation; H5N1; Influenza virus; Neuraminidase; TCM
Download this article as:| Copy the following to cite this article: Odhar H. A, Ibrahim A. A, Hashim A. F. Computational Screening of Traditional Chinese Medicine (TCM) Library to Identify Potential Inhibitors of H5N1 Avian Influenza Neuraminidase: A Molecular Docking and Dynamics Simulation Study. Biomed Pharmacol J 2025;18(2). |
| Copy the following to cite this URL: Odhar H. A, Ibrahim A. A, Hashim A. F. Computational Screening of Traditional Chinese Medicine (TCM) Library to Identify Potential Inhibitors of H5N1 Avian Influenza Neuraminidase: A Molecular Docking and Dynamics Simulation Study. Biomed Pharmacol J 2025;18(2). Available from: https://bit.ly/3FLraZh |
Introduction
Influenza is an infective disease that mainly affect the upper respiratory tract, leading to remarkable mortality and morbidity in human population during seasonal epidemics and occasional pandemics.1 Annually, there are one billion cases of seasonal influenza worldwide. Of these global influenza cases, about 3-5 million patients can develop severe respiratory illness with estimates of 290,000 to 650,000 deaths.2 The symptoms of influenza infection can vary from mild to severe, these symptoms generally include: fever, muscles pain, chills, cough, headache, sore throat, pneumonia and sometimes death. The severe cases of influenza are generally reported in high risk patients like children, elderly, pregnant women and immunocompromised individuals.3 In human beings, Influenza disease is usually caused by influenza A or B viruses. Moreover, the influenza C virus can cause mild cases of the disease and represents a less public health threat. Among these three types of influenza virus, the greatest threat to human health is usually imposed by influenza type A virus due to its known tendency for antigenic variation and its capacity to drive a pandemic.4
Influenza virus is a negative-sense and single stranded RNA pathogen, this virus has two antigenic glycoproteins on its surface: hemagglutinin (HA) and neuraminidase (NA). These two glycoproteins are critical to influenza virus entry and release from host cells. Based on the serology of HA and NA glycoproteins, the influenza A virus can be classified into several subtypes like H1N1, H2N2, H3N2 and H5N1.5 It is believed that viral genome segmented nature and lack of proof-reading capacity in RNA-dependent RNA polymerase makes influenza virus highly mutagenic and susceptible for both antigenic shift and drift. Therefore, the development of a long-lasting influenza vaccine is still challenging.6 Fortunately, the antiviral agents are considered another defensive tool against the rapidly evolving influenza virus or any novel strain that may arise in future. The currently FDA approved antiviral drugs for treatment of influenza infection are categorized into two main groups: the M2 ion channel inhibitors and the neuraminidase inhibitors (NAIs).1 The M2 ion channel inhibitors like amantadine and rimantadine are only effective against influenza A virus, these two antivirals can inhibit viral disassembly inside host cells. However, this group of antiviral drugs are no longer recommended for treatment of influenza A virus infection as the currently circulating strains are fully resistant to them.7 On the other hand, the NAIs like zanamivir, oseltamivir and peramivir are still active against the circulating strains of both influenza A and B viruses despite the emergence of resistance in some instances. The mechanism of action for NAIs involves binding of these agents to the viral NA glycoprotein, the competitive inhibition of NA will prevent the cleavage of N-acetyl neuraminic acid in host cells and thereby preventing the release of new virions.8 However, the emergence of some resistant strains to NAIs like the oseltamivir resistant H1N1 strain (H274Y mutant) necessitates the development of novel NAIs to overcome this challenge.9 Recently, an outbreak of a pathogenic avian influenza H5N1 was reported among cattle herds in the United States. Few farm workers were also infected but more cases may have gone undetected. Nowadays, there are concerns that the virus may undergo evolution to easily infect other mammals including human.10 Therefore, in this study, we have virtually screened natural compounds in the Traditional Chinese Medicine (TCM) library to identify probable inhibitors of H5N1 avian influenza neuraminidase glycoprotein. The aim of this computational study is to employ both docking and dynamics simulation in order to identify a lead compound towards the development of inhibitor against H5N1 avian influenza neuraminidase.
Materials and Methods
Setting up virtual screening plan
An outline for the major steps of this virtual screening study can be viewed in Figure 1. Briefly, the first step of this study involved docking of the compounds in the TCM library against the H5N1 influenza virus neuraminidase. Then, the best ten compounds in the docking output with the least energy of binding were subjected to the next step. In the second step of this study, these top ten compounds were submitted to molecular dynamics (MD) simulation for 25 nanoseconds. In this initial round of MD simulation, the ligand movement was reported as root mean square deviation (RMSD). As such, only those compounds with a mean ligand movement RMSD less than 4 Angstrom were submitted to the second round of MD simulation for 50 nanoseconds. Finally, the final hits were selected from the second simulation based on both mean ligand movement RMSD (less than 4 Angstrom) and average molecular mechanics-Poisson Boltzmann surface area (MM-PBSA) binding energy.
 |
Figure 1: A schematic representation for the major stages of this in silico study.Click here to view Figure |
Molecular Docking
In this step, we have used DrugRep online server to carry out the molecular docking of compounds in the TCM library against H5N1 influenza virus neuraminidase.11 At first, chain B of neuraminidase crystal (PDB: 2HU0) was uploaded to the DrugRep server.12 It is worth mentioning that this chain B was extracted from the tetrameric crystal by using UCSF Chimera version 1.15.13 Also, the co-crystalized ligand (oseltamivir carboxylate) was removed from chain B crystal by using the aforementioned software. During docking step, the applied coordinates were X: -2.0, Y: 23.0, Z: 110.0 while the grid box size was 20*20*20 Angstrom. Moreover, the DrugRep server employed both AutoDock Vina version 1.1.2 and AutoDockTools version 1.5.6 to accomplish the structure-based virtual screening.14,15 Additionally, the precision of docking procedure was evaluated by redocking the co-crystalized oseltamivir carboxylate into the neuraminidase chain B. Then, the PacDOCK web server was used to calculate the degree of conformations alignment between co-crystalized and docked ligands.16 When the docking step was completed, we have selected only the top ten compounds with least energy of binding for additional study. For these top compounds, the orientation with a minimum binding energy pose for each docking complex was visualized by PyMOL version 2.4.1 and Discovery Studio Visualizer version 21.1.0.17,18 In addition, different chemical and pharmacokinetics features for these top hits were predicted by utilizing several webservers like: pkCSM, SwissADME, Molsoft L.L.C. and ProTox 3.0.19–22 Finally, these top ten compounds from the docking results were subjected to the next step of molecular dynamics (MD) simulation.
Molecular dynamics (MD) simulation study
In this study, the software YASARA Dynamics version 20.12.24 was used to execute two successive rounds of MD simulation for 25 and 50 nanoseconds respectively.23 For each of the top docking hits, the ligand-neuraminidase complex with a minimum binding energy pose was subjected for 25 nanoseconds MD simulation at first. During this simulation, the ligand proximity to neuraminidase active site was reported as RMSD value. As such, only those compounds with a mean ligand proximity RMSD of less than 4 Angstrom were then subjected to undergo the second round of 50 nanoseconds simulation. Finally, the best hits were selected based on the calculated mean ligand proximity RMSD that didn’t exceed 4 Angstrom. Moreover, the average molecular mechanics-Poisson Boltzmann surface area (MM-PBSA) binding energy was calculated by applying AMBER14 force field.24 For comparative purpose, the docking complex between oseltamivir carboxylate and neuraminidase was subjected to the same two rounds of MD simulation. The reported results for the MD simulation of oseltamivir carboxylate-neuraminidase complex were used as positive control in this study. The procedure applied to operate this MD simulation is similar in details to what we have used in our previous studies.25–27 In summary, a concentration of 0.9% sodium chloride was used in this simulation. An additional concentration of either sodium or chloride ions was added for the neutralization of ligand-neuraminidase complex. The following forcefields were applied in this simulation study: AMBER14 for the solute, TIP3P for water, AM1BCC and GAFF2 for the ligand.24,28,29
Results
At first, the reliability of applied docking protocol was evaluated by using the redocking method. In this method, the co-crystalized oseltamivir carboxylate was removed from neuraminidase chain B. Then, the oseltamivir carboxylate was docked again into the neuraminidase active site by applying the same docking protocol used in this study. After that, the conformation of the co-crystalized oseltamivir carboxylate was aligned with the docked one. The degree in conformation difference between the native and docked oseltamivir carboxylate was reported as RMSD value. In this study, the conformation alignment between co-crystalized and docked oseltamivir carboxylate can be seen in Figure 2. As indicated in this figure, the calculated conformation difference RMSD was 1.538 Angstrom. Also, it is worth to mention that the energy of binding for redocking oseltamivir carboxylate was 6.1 Kcal/ mol.
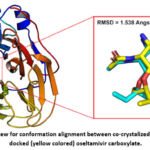 |
Figure 2: An overview for conformation alignment between co-crystalized (cyan colored) and docked (yellow colored) oseltamivir carboxylate.Click here to view Figure |
A summary is given in Table 1 for the botanical sources and pharmacological effects of the best ten hit compounds identified by the docking study. In addition, various chemical characteristics are shown in Table 2 for the best ten compounds obtained by the structure-based virtual screening study. As seen in these tables, the top compounds were listed based on their minimum energy of binding (docking energy) against chain B of neuraminidase crystal. When considering the chemical formula of these listed compounds, we can recognize that all these compounds are flavonoids (polyphenolic) with the exception of compound number 8. The hit number 8, isoliensinine, is an alkaloid nitrogen-based compound with less hydrogen bond accepting or donating groups. Moreover, all the listed compounds in Table 2 have a logarithm of octanol/ water partition coefficient (Log P) that is lower than 5 with the exception of compound number 8. Thus, isoliensinine can be considered less hydrophilic as compared to the other listed compounds.
Table 1: A summary list for botanical sources and pharmacological activities of the top hit compounds in this virtual screening study.
| No. | Hit name | Source | Activity |
| 1 | Grosvenorine | Siraitia grosvenorii | Antibacterial, antioxidant.30 |
| 2 | Amentoflavone | Selaginella tamariscina | Anti-inflammatory, anticancer, antimicrobial.31 |
| 3 | Methyl hesperidin | Plantago depressa, Citrus reticulata, Citrus deliciosa | Vasodilation.32 |
| 4 | Bilobetin | Ginkgo biloba | Decreases blood lipids, enhances effect of insulin.33 |
| 5 | Eriocitrin | Citrus sulcata, Citrus reticulata | Antioxidant, anticancer.34 |
| 6 | Pectolinarin | Cirsium japonicum | Anti-inflammatory.35 |
| 7 | Linarin | Mentha arvensis | Acetylcholinesterase inhibitor.36 |
| 8 | Isoliensinine | Nelumbo nucifera | Anti-inflammatory, antioxidant, anticancer.37 |
| 9 | Tiliroside | Daphne genkwa, Leonurus japonicus | Anti-diabetic.38 |
| 10 | Robinin | Robinia pseudoacacia, Vinca erecta | Anti-inflammatory, anticancer.39 |
Table 2: Chemical properties for the top ten hits generated by the virtual screening of traditional Chinese medicine (TCM) library against chain B of N1 neuraminidase crystal. These hits were ranked based on their least docking energy to target crystal.
| No. | Compound name | Chemical formula | Docking score (Kcal/ mol) | M.W. (g/mol) | HBD | HBA | Log P |
| 1 | Grosvenorine | C33H40O19 | -10.0 | 740.70 | 11 | 12 | -2.2 |
| 2 | Amentoflavone | C30H18O10 | -9.8 | 538.46 | 6 | 8 | 3.4 |
| 3 | Methyl hesperidin | C29H36O15 | -9.7 | 624.59 | 7 | 8 | -0.7 |
| 4 | Bilobetin | C31H20O10 | -9.7 | 552.49 | 5 | 7 | 3.7 |
| 5 | Eriocitrin | C27H32O15 | -9.3 | 596.53 | 9 | 10 | -1.4 |
| 6 | Pectolinarin | C29H34O15 | -9.3 | 622.59 | 7 | 8 | -1.2 |
| 7 | Linarin | C28H32O14 | -9.2 | 592.55 | 7 | 8 | -1.2 |
| 8 | Isoliensinine | C37H42N2O6 | -9.2 | 610.75 | 2 | 2 | 6.3 |
| 9 | Tiliroside | C30H26O13 | -9.1 | 594.52 | 7 | 9 | 1.8 |
| 10 | Robinin | C33H40O19 | -9.0 | 740.66 | 11 | 12 | -2.8 |
M.W.: molecular weight; HBD: hydrogen bond donor; HBA: hydrogen bond acceptor; Log P: logarithm of partition coefficient.
A prediction list of drug-likeness score, pharmacokinetics and toxicity characteristics can be seen in Table 3 for the top ten compounds. Again, these best compounds in Table 3 were arranged according to their least docking energy to neuraminidase crystal. When considering drug-likeness feature, all the listed compounds have a relatively high score except amentoflavone and bilobetin (compounds number 2 and 4). These two compounds, together with compound number 8 (isoliensinine), are predicted to have poor solubility in water and low volume of distribution. Additionally, all compounds in Table 3 are predicted to have minimal intestinal absorption except compound number 8 possibly due to the high Log P value for this compound. Finally, all the compounds in Table 3 are considered to be non-mutagenic based on AMES toxicity prediction. However, compounds number 2, 4 and 8 are expected to have the least median lethal dose (LD50) when compared to the other hits.
Table 3: A tabular summary for drug-likeness score, pharmacokinetics and toxicity parameters of top hit compounds. These hits were listed based on their least docking energy against N1 neuraminidase chain B.
| No. | Hit name | Drug-likeness | Pharmacokinetics | Toxicity | |||
| Water solubility (mg/ml) | Intestinal absorption | VDss(L/Kg) | AMES toxicity | LD50(mg/ Kg) | |||
| 1 | Grosvenorine | 0.83 | 5.65e-01 (soluble) | Low | 10.23 | No | 5000 |
| 2 | Amentoflavone | 0.19 | 9.63e-05(poorly soluble) | Low | 0.08 | No | 3919 |
| 3 | Methyl hesperidin | 1.00 | 6.98e-01(soluble) | Low | 3.81 | No | 12000 |
| 4 | Bilobetin | 0.27 | 6.12e-05(poorly soluble) | Low | 0.07 | No | 4000 |
| 5 | Eriocitrin | 1.13 | 1.87e+00(soluble) | Low | 54.20 | No | 12000 |
| 6 | Pectolinarin | 0.87 | 1.14e-01(soluble) | Low | 4.83 | No | 5000 |
| 7 | Linarin | 0.74 | 1.33e-01(soluble) | Low | 8.30 | No | 5000 |
| 8 | Isoliensinine | 1.71 | 2.18e-05(poorly soluble) | High | 0.17 | No | 1180 |
| 9 | Tiliroside | 0.75 | 6.94e-03(moderately soluble) | Low | 4.52 | No | 5000 |
| 10 | Robinin | 0.86 | 3.50e-01(soluble) | Low | 14.06 | No | 5000 |
VDss: steady state volume of distribution; LD50: median lethal dose.
In the final stage of this computation study, the best ten hit compounds were subjected to MD simulation study for two consecutive rounds of 25 and 50 nanoseconds. The overall results of these two rounds were presented in Table 4. As observed in this table, the proximity of each hit compound to neuraminidase active site was calculated as a mean value for ligand movement RMSD. As shown in Table 4, only five compounds were able to record mean ligand movement RMSD value of less than 4 Angstrom during 30 nanoseconds simulation interval. Then, these five hit compounds were subjected to the second round of MD simulation for 50 nanoseconds. Throughout this second round, only grosvenorine and pectolinarin were able to keep a low mean ligand movement RMSD of 3.97 and 2.63 Angstrom respectively. As compared to other hits in Table 4, the compound pectolinarin had the least average MM-PBSA binding energy of -115.24 Kcal/mol. A detailed plot for the proximity of grosvenorine, pectolinarin and oseltamivir carboxylate to neuraminidase active site can be seen in Figure 3 during 50 nanoseconds period. Unlike grosvenorine and pectolinarin, it is very clear that the ligand oseltamivir carboxylate had started moving away from neuraminidase active site after 22 nanoseconds of simulation as presented in Figure 3.
Table 4: A summarized results for molecular dynamics (MD) simulation of the best hits.
| No. | Compound name | MD simulation interval | ||
| 20 nanoseconds | 50 nanoseconds | |||
| Mean ligand movement RMSD (Å) | Mean ligand movement RMSD (Å) | Average MM-PBSA binding energy (Kcal/ mol) | ||
| 1 | Grosvenorine | 3.71 | 3.97 | -90.95 |
| 2 | Amentoflavone | 6.26 | – | – |
| 3 | Methyl hesperidin | 3.60 | 5.33 | -56.857 |
| 4 | Bilobetin | 11.98 | – | – |
| 5 | Eriocitrin | 10.84 | – | – |
| 6 | Pectolinarin | 2.49 | 2.63 | -115.24 |
| 7 | Linarin | 6.94 | – | – |
| 8 | Isoliensinine | 7.11 | – | – |
| 9 | Tiliroside | 3.08 | 7.46 | -44.26 |
| 10 | Robinin | 3.56 | 4.32 | -100.73 |
| 11 | Oseltamivir carboxylate | 3.36 | 4.96 | -60.88 |
MD: Molecular dynamics; RMSD: Root mean square deviation; Å: Angstrom; MM-PBSA: Molecular mechanics-Poisson Boltzmann surface area.
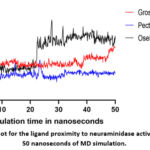 |
Figure 3: A plot for the ligand proximity to neuraminidase active site during 50 nanoseconds of MD simulation.Click here to view Figure |
For the current in-silico study, the two and three-dimensional images for the docking of grosvenorine and pectolinarin against neuraminidase are presented in Figure 4. It is evident that grosvenorine is involved in five interactions with active site key residues of neuraminidase as seen in Figure 4 (A), while pectolinarin is only engaged in two interactions as illustrated in Figure 4 (B). The list of neuraminidase active site key residues that are involved in chemical interactions with docked grosvenorine and pectolinarin is presented in Table 5.
Table 5: Key residues of neuraminidase active site that are engaged in interactions with grosvenorine and pectolinarin.
| Ligand | Active site residues | ||||||||
| Arg118 | Asn146 | Asp151 | Arg152 | Arg224 | Glu276 | Arg292 | Arg371 | Tyr406 | |
| Grosvenorine | – | – | Yes | – | Yes | Yes | – | Yes | Yes |
| Pectolinarin | – | – | Yes | – | Yes | – | – | – | – |
 |
Figure 4: Docking images for (A) grosvenorine and (B) pectolinarin against neuraminidase chain B.Click here to view Figure |
Discussion
The influenza virus is a single stranded RNA microorganism with a mutagenic and pandemic potential. Currently, the pandemic threat of influenza is highly probable due to the recent outbreak of avian influenza H5N1 among cattle herds in the United States. It is possible that this influenza strain may undergo evolution that enable the virus to cross species barrier and infect other mammals like human.10 Therefore, it was of our interest to computationally screen a library of natural compounds against N1 glycoprotein of influenza virus. The aim of this in-silico study is to find a potential inhibitor for the avian influenza H5N1 strain.
At first, the accuracy of the docking protocol was assessed in this virtual screening study. For this purpose, the redocking approach was employed as seen in Figure 2 of the results part. According to Figure 2, the conformations difference RMSD was 1.538 Angstrom between co-crystalized and docked oseltamivir carboxylate. As such, this low RMSD value means that the used docking protocol in this study is reliable and has a good accuracy. It is well-known that low RMSD value refers to less degree of conformation change between the native and docked ligands, and thus reflects good docking protocol accuracy. Based on published articles, a conformation difference RMSD ranging between 1.5 and 2.0 Angstrom usually point to good docking accuracy.40
When considering the predicted chemical features, it is obvious that all the listed compounds in Table 2 don’t comply with Lipinski’s rule of five 41 as all these compounds have a molecular weight greater than 500 g/ mol. Also, many of these hits have hydrogen bond donating groups greater than 5. Therefore, it is predicted that these listed compounds in Table 2 may have poor oral bioavailability. Interestingly, the anticipated Log P values for amentoflavone, bilobetin and isoliensinine were higher than those listed for other hits in Table 2. These predicted values for partition coefficient parameter in Table 2 may refer to the more hydrophobic nature of these three compounds and also explain their expected pharmacokinetics behavior in Table 3. According to the Table 3 findings, it is anticipated that the three compounds with list number 2, 4 and 8 don’t have favorable pharmacokinetics and toxicological features toward the development of potential lead compound.
In the analysis of MD results, it is well-known that less ligand movement RMSD refers to closer proximity of the ligand to target active site, thus stronger binding can be deduced. For this measurement, the ligand movement RMSD parameter was calculated by superimposing the compound-neuraminidase complex on its reference structure throughout simulation interval.42 The MD simulation results in Figure 3 and Table 4 suggest that both grosvenorine and pectolinarin may be superior in binding target crystal than other studied compounds, including oseltamivir carboxylate.
Finally, the examination of docking images can identify the possible interactions involved in the binding between target active site residues and studied ligands. It is well-known that the enzyme neuraminidase is a homo-tetramer, the catalytic active site is located on the head of each tetramer in a negatively charged pocket. This active site is made up of the following conserved amino acid residues: Arginine 118, Asparagine 146, Aspartic acid 151, Arginine 152, Arginine 224, Glutamic acid 276, Arginine 292, Arginine 371, Tyrosine 406.43 Based on these facts, we can recognize that both grosvenorine and pectolinarin were involved in several chemical interactions with neuraminidase active site key residues as illustrated in Figure 4 (A) and (B) respectively.
Limitations of this computational study are numerous like the influence of Vina scoring function on the accuracy of ligand binding modes in the docking results. It is a known fact that the software AutoDock Vina employs a united-atom scoring function that relies only on the heavy atoms rather than hydrogen atoms which can adversely affect docking results. However, the accuracy of the used scoring function is considered target specific in general and it should be assessed if possible, by redocking the native ligand to target active site.14,44 Another possible limitation in this study is the neglection of entropy contribution to binding free energy estimation by MM-PBSA method due to the extensive computational cost of this approach.45 As such, the contribution of solvation entropy to the binding energy is not represented in our study. Therefore, it is advisable to verify the results of this in-silico study both in-vitro and in-vivo settings.
Conclusion
In this in-silico study, the Traditional Chinese Medicine (TCM) database was screened against influenza virus H5N1 neuraminidase monomer by using molecular docking and dynamics simulation. According to the results of this study, both grosvenorine and pectolinarin may have the potential to inhibit the neuraminidase glycoprotein and thereby impeding the release of new virions from infected cells. Based on docking analysis, both compounds are expected to be involved in numerous interactions with neuraminidase active site key residues. Moreover, these two hit compounds were able to keep a close ligand proximity to target active site in molecular dynamics simulation. Interestingly, the flavonoid pectolinarin has the least average molecular mechanics-Poisson Boltzmann surface area binding energy during simulation. Additionally, both grosvenorine and pectolinarin are predicted to be hydrophilic with low intestinal absorption and high median lethal dose. However, these computational results must be further assessed both in-vitro and in-vivo.
Acknowledgment
The authors would like to thanks the department of pharmacy, Al-Zahrawi University College for their support of this work.
Funding Source
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Permission to reproduce material from other sources
Not Applicable.
Clinical Trial Registration
This research does not involve any clinical trials.
Authors’ Contribution
- Hasanain Abdulhameed Odhar: Conceptualization, Methodology, Data collection, Analysis, Writing – Original Draft.
- Azher Abdulmutaleb Ibrahim: Writing – Review & Editing, Supervision.
- Ahmed Fadhil Hashim: Writing – Review, Supervision.
Reference
- Han J, Perez J, Schafer A, et al. Influenza Virus: Small Molecule Therapeutics and Mechanisms of Antiviral Resistance. Curr Med Chem. 2018;25(38):5115.
CrossRef - Influenza (Seasonal). Accessed July 11, 2024. https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal)
- Keilman LJ. Seasonal Influenza (Flu). Nurs Clin North Am. 2019;54(2):227-243.
CrossRef - Świerczyńska M, Mirowska-Guzel DM, Pindelska E. Antiviral Drugs in Influenza. Int J Environ Res Public Health. 2022;19(5).
CrossRef - Bouvier NM, Palese P. THE BIOLOGY OF INFLUENZA VIRUSES. Vaccine. 2008;26(Suppl 4):D49.
CrossRef - Ahmed R, Oldstone MBA, Palese P. Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat Immunol. 2007;8(11):1188.
CrossRef - Balannik V, Wang J, Ohigashi Y, et al. Design and Pharmacological Characterization of Inhibitors of Amantadine-Resistant Mutants of the M2 Ion Channel of Influenza A Virus. Biochemistry. 2009;48(50):11872.
CrossRef - Mckimm-Breschkin JL. Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza Other Respi Viruses. 2013;7(Suppl 1):25.
CrossRef - Butler J, Hooper KA, Petrie S, et al. Estimating the Fitness Advantage Conferred by Permissive Neuraminidase Mutations in Recent Oseltamivir-Resistant A(H1N1)pdm09 Influenza Viruses. PLoS Pathog. 2014;10(4).
CrossRef - Mallapaty S. Bird flu could become a human pandemic. How are countries preparing? Nature. Published online July 12, 2024.
CrossRef - Gan J hong, Liu J xiang, Liu Y, et al. DrugRep: an automatic virtual screening server for drug repurposing. Acta Pharmacol Sin 2022 444. 2022;44(4):888-896.
CrossRef - Russell RJ, Haire LF, Stevens DJ, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443(7107):45-49.
CrossRef - Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-1612.
CrossRef - Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31(2):455.
CrossRef - Morris GM, Ruth H, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30(16):2785.
CrossRef - Carbone J, Ghidini A, Romano A, Gentilucci L, Musiani F. PacDOCK: A Web Server for Positional Distance-Based and Interaction-Based Analysis of Docking Results. Molecules. 2022;27(20):6884.
CrossRef - PyMOL | pymol.org. Accessed July 17, 2024. https://pymol.org/
- BIOVIA Discovery Studio Visualizer | Dassault Systèmes. Accessed July 17, 2024. https://www.3ds.com/products/biovia/discovery-studio/visualization
- pkCSM. Accessed July 17, 2024. https://biosig.lab.uq.edu.au/pkcsm/prediction
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7.
CrossRef - Molsoft L.L.C.: Drug-Likeness and molecular property prediction. Accessed July 17, 2024. https://molsoft.com/mprop/
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(Web Server issue):W257.
CrossRef - Krieger E, Vriend G. YASARA View – molecular graphics for all devices – from smartphones to workstations. Bioinformatics. 2014;30(20):2981-2982.
CrossRef - Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157-1174.
CrossRef - Odhar HA, Hashim AF, Ahjel SW, Humadi SS. Molecular docking and dynamics simulation analysis of the human FXIIa with compounds from the Mcule database. Bioinformation. 2023;19(2):160.
CrossRef - Odhar HA, Hashim AF, Humadi SS, Ahjel SW. LIGAND-BASED VIRTUAL SCREENING OF FDA-APPROVED DRUGS TO IDENTIFY NEW INHIBITORS AGAINST LACTATE DEHYDROGENASE ENZYME OF MALARIA PARASITES. Int J Appl Pharm. 2024;16(1):255-260.
CrossRef - Odhar HA, Hashim AF, Ahjel SW, Humadi SS. VIRTUAL SCREENING OF FDA-APPROVED DRUGS BY MOLECULAR DOCKING AND DYNAMICS SIMULATION TO RECOGNIZE POTENTIAL INHIBITORS AGAINST MYCOBACTERIUM TUBERCULOSIS ENOYL-ACYL CARRIER PROTEIN REDUCTASE ENZYME. Int J Appl Pharm. 2024;16(1):261-266.
CrossRef - Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696.
CrossRef - Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23(16):1623-1641.
CrossRef - Wang M, Xing S, Luu T, Fan M, Li X. The Gastrointestinal Tract Metabolism and Pharmacological Activities of Grosvenorine, a Major and Characteristic Flavonoid in the Fruits of Siraitia grosvenorii. Chem Biodivers. 2015;12(11):1652-1664.
CrossRef - Xiong X, Tang N, Lai X, et al. Insights Into Amentoflavone: A Natural Multifunctional Biflavonoid. Front Pharmacol. 2021;12.
CrossRef - Kikuchi K, Hirata M, Imai Y, Aramaki Y. The potentiation of the coronary dilating and the cardiac action of adenosine or adenine nucleotides by methylhesperidin. Jpn J Pharmacol. 1966;16(2):224-225.
CrossRef - Kou XH, Zhu MF, Chen D, et al. Bilobetin ameliorates insulin resistance by PKA-mediated phosphorylation of PPARα in rats fed a high-fat diet. Br J Pharmacol. 2012;165(8):2692-2706.
CrossRef - Wang Z, Zhang H, Zhou J, et al. Eriocitrin from lemon suppresses the proliferation of human hepatocellular carcinoma cells through inducing apoptosis and arresting cell cycle. Cancer Chemother Pharmacol. 2016;78(6):1143-1150.
CrossRef - Wang L, Wang N, Zhao Q, Zhang B, Ding Y. Pectolinarin inhibits proliferation, induces apoptosis, and suppresses inflammation in rheumatoid arthritis fibroblast-like synoviocytes by inactivating the phosphatidylinositol 3 kinase/protein kinase B pathway. J Cell Biochem. 2019;120(9):15202-15210.
CrossRef - Oinonen PP, Jokela JK, Hatakka AI, Vuorela PM. Linarin, a selective acetylcholinesterase inhibitor from Mentha arvensis. Fitoterapia. 2006;77(6):429-434.
CrossRef - Cheng Y, Li HL, Zhou ZW, et al. Isoliensinine: A Natural Compound with “Drug-Like” Potential. Front Pharmacol. 2021;12.
CrossRef - Goto T, Horita M, Nagai H, et al. Tiliroside, a glycosidic flavonoid, inhibits carbohydrate digestion and glucose absorption in the gastrointestinal tract. Mol Nutr Food Res. 2012;56(3):435-445.
CrossRef - Zhang W, Liu W, Hu X. Robinin inhibits pancreatic cancer cell proliferation, EMT and inflammation via regulating TLR2-PI3k-AKT signaling pathway. Cancer Cell Int. 2023;23(1).
CrossRef - Hevener KE, Zhao W, Ball DM, et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model. 2009;49(2):444-460.
CrossRef - Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3-26.
CrossRef - Odhar HA, Ahjel SW, Albeer AAMA, Hashim AF, Rayshan AM, Humadi SS. Molecular docking and dynamics simulation of FDA approved drugs with the main protease from 2019 novel coronavirus. Bioinformation. 2020;16(3):236-244.
CrossRef - McAuley JL, Gilbertson BP, Trifkovic S, Brown LE, McKimm-Breschkin JL. Influenza Virus Neuraminidase: Structure and Function. Acta Naturae. 2009;1(2):26.
CrossRef - Odhar HA, Odhar ZA, Muhi MR. Screening of Traditional Chinese Medicine Library Against Penicillin-binding Protein 2a for Methicillin-resistant Staphylococcus aureus by Molecular Docking, Dynamics Simulation and In vitro Antimicrobial Activity. Biomed Pharmacol J. 2025;18(1):823-834.
CrossRef - Dong L, Li P, Wang B. Enhancing MM/P(G)BSA Methods: Integration of Formulaic Entropy for Improved Binding Free Energy Calculations. J Comput Chem. 2025;46(10):e70093.
CrossRef
List of abbreviations
A: Angstrom.
HA: Hemagglutinin.
HBA: Hydrogen bond acceptor.
HBD: Hydrogen bond donor.
LD50: Median lethal dose.
Log P: Logarithm of partition coefficient.
M.W.: Molecular weight.
MD: Molecular dynamics.
MM-PBSA: Molecular mechanics-Poisson Boltzmann surface area.
NA: Neuraminidase.
NAI: Neuraminidase inhibitor.
RMSD: Root mean square deviation.
TCM: Traditional Chinese Medicine.
VDss: Steady state volume of distribution.

