
Manuscript accepted on :21-04-2025
Published online on: 07-05-2025
Plagiarism Check: Yes
Reviewed by: Dr. Mohammed Ahmed
Second Review by: Dr. Priya Bhardwaj
Final Approval by: Dr. Mariia Shanaida
Raje Siddiraju Upendra* , Mattur Srinivasa Murthy Upamanyu, Sanjay Shrinivas Nagar, Preetham Rechihalli Shivalingappa, Sandeep Anjaneya and Karthik Rajendra
, Mattur Srinivasa Murthy Upamanyu, Sanjay Shrinivas Nagar, Preetham Rechihalli Shivalingappa, Sandeep Anjaneya and Karthik Rajendra
School of Electronics and Communication Engineering, REVA University, Bangalore, India
Corresponding Author E-mail: upendra.rs@reva.edu.in
DOI : https://dx.doi.org/10.13005/bpj/3178
Abstract
Influenza, a highly contagious respiratory infection, continues to threaten global health with significant transmission rates and annual fatalities of 400,000. The virus's rapid mutation and genetic reassortment necessitate the development of effective antiviral drugs. Among other proteins related to the influenza virus, Hemagglutinin (HA) helps the virus to enter and invade the human body. Hence in the present study molecular docking combined with pharmacokinetic assessments was implemented to streamline drug development for the target protein Hemagglutinin (HA). A database of 24 potential compounds was screened using PubChem, followed by ADMET and drug-likeness analysis. A total of 07 ligands retrieved from PubChem database with favorable profiles were considered for the molecular docking process with hemagglutinin as the target protein through the AutoDock Vina package present in the PyRx software. The docking results identified that among the tested compounds, 5,7,4’-Trihydroxy-3-methoxyflavanone isolated from plant Populus szechuanica and retrieved from PubChem database (PubChem ID 72281) exhibited the highest docking score of -8.5 kcal/mol in comparison with standard drug Apigenin which displayed a docking score of -8.4 kcal/mol against target protein HA. Hence compound 5,7,4’-Trihydroxy-3-methoxyflavanone can be further developed as an effective drug against the influenza virus.
Keywords
Apigenin; Docking; Hemagglutinin; Influenza; 5,7,4’-Trihydroxy-3-Methoxyflavanone
Download this article as:| Copy the following to cite this article: Upendra R. S, Upamanyu M. S. M, Nagar S. S, Shivalingappa P. R, Anjaneya S, Rajendra K. Computational and Experimental Discovery of Hemagglutinin-Targeting Agents from Populus szechuanica: Molecular Docking, Characterization, and Antiviral Potential. Biomed Pharmacol J 2025;18(2). |
| Copy the following to cite this URL: Upendra R. S, Upamanyu M. S. M, Nagar S. S, Shivalingappa P. R, Anjaneya S, Rajendra K. Computational and Experimental Discovery of Hemagglutinin-Targeting Agents from Populus szechuanica: Molecular Docking, Characterization, and Antiviral Potential. Biomed Pharmacol J 2025;18(2). Available from: https://bit.ly/4iS48h1 |
Introduction
Influenza, sometimes referred to as the flu, is an extremely infectious respiratory infection caused by influenza viruses. The influenza virus still poses a danger to world health despite advances in medical technology, resulting in yearly outbreaks and sporadic pandemics. Between 13 March and 20 June 2024, 14 new human cases of avian influenza virus infection were reported worldwide.1 A gradient rise in influenza cases was observed during COVID-19 infection due to decrease in immunity caused by SARS-CoV-2 virus. Hence there is need to study phytocompounds to develop strong medication against influenza.2 The ever-evolving nature of influenza viruses, marked by genetic reassortment and mutations, makes the development of potent antiviral medications imperative. Many potential target proteins were studied to discover novel drugs against the influenza virus. Among these target proteins, the protein HA was considered for the present study due to its significant role in assisting the entry of viruses into the human body.3 Hence HA was selected as the target protein in the present study to find novel drugs with the help of computational biology. Combining molecular docking studies with pharmacokinetic tests within this framework appears to be a viable way to accelerate drug development. With the help of computational biology, many phytocompounds isolated from different plants (Toddalia asiatica,4 Curcuma aromatica,5 Selaginella tamariscina6) could be investigated as novel drugs against the influenza virus. Scientists can forecast a drug’s binding affinity with the target protein by modeling the binding of putative antiviral drugs to particular viral protein targets. The ligand or chemical compounds tested for inhibitory effect against target protein HA were retrieved from PubChem database using molecular docking and ADMET studies.
Materials and Methods
PubChem screening of potential compounds
With the standard drugs apigenin, 7 and isorhapontigenin, 8 as the input, data mining was performed in the PubChem database to obtain the chemical compounds displaying properties similar to the input compound through the “2D Tanimoto Similarity Search” package.9 After the completion of the data mining process, the output molecules similar to the input molecule were filtered through Lipinski’s Rule of Five (i.e. octanol-water partition coefficient (logP) < 5, molecular weight < 500Da, hydrogen bond acceptors ≤ 10, and hydrogen bond donors ≤ 5), 10,11 to obtain the molecules with more potential of become lead molecules. At the end of the filtering process, molecules displaying Lipinski’s Rule of Five were saved for druglikeness and ADMET studies.12
ADMET and Druglikeness analysis of database
SMILE format of the compounds present in the Excel file prepared at the end of PubChem screening was imported into Drulito software for ADMET and druglikeness test.13 All the selected 800 compounds were tested for both ADMET and druglikeness properties such as BBB, Ghosh filter, CMC 50-like rule, Veber filter, MDDR-like rule, uwQED, and wQED. The output generated was saved as an Excel file and analyzed with the help of Python script to filter out the compounds exhibiting ADMET and druglikeness results within the accepted range were further selected as the ligand molecules for the docking process for the target protein HA.14
Target protein preparation in UCSF Chimera
UCSF Chimaera software was utilized to line up the target protein that was chosen for molecular docking. The protein molecule HA with PDB ID 3AL4’s15 3D structure was obtained from the PDB database and fed into UCSF Chimaera to produce the protein. Initially, all the water molecules were removed from the protein structure, and hydrogen bonds (polar and non-polar) and charges (Kollman) were added to the dehydrated protein structure to facilitate proper molecular docking with ligand molecules using the Dockprep tool present in the UCSF Chimera. The constructed protein structure was given the appropriate grid box and stored in PDBQT format for docking.16
Ligand preparation for molecular docking
Using PyRx software, all the chosen ligand molecules as well as the standard drug were ready for the molecular docking procedure.17 All the ligand molecules were entered into the PyRx application as input for molecular docking after being downloaded in SDF format from the PubChem database. Initially, energy minimization was performed to ensure the stability of the ligand molecules, which plays a crucial role in the molecular docking process. Stabilized ligand molecules were then given additional hydrogen bonds and charge, and the final structure was recorded in PDBQT format for the molecular docking process.18
Molecular docking of ligands with hemagglutinin
Both ligand molecules and target protein in PDBQT format were processed as input into PyRx software for the molecular docking process.19 After providing input, “AutoDock wizard” was implemented for molecular docking, and necessary inputs such as ligand molecule names, target protein names, grid box coordinates, and the number of exhaustiveness were provided to ensure a site-specific molecular docking process. The output generated was saved as an SDF file of docked ligand molecules and further provided as the input to form the complex and study docking between protein and ligand molecules.20
Protein-ligand analysis in Discovery Studio
SDF files generated as the output of the docking process were provided as input for protein-ligand interaction analysis in Discovery Studio Software. 21 After providing the input SDF file, the final structure of the target protein was merged with the SDF file to form a protein-ligand complex. PDB format of protein-ligand complex was stored in PDB format and the “2D diagram” option was used to visualize the interaction between ligand molecule and target protein hemagglutinin in 2D diagram format. This 2D interaction image was saved in image format for documentation purposes. The entire protocol was repeated for all selected ligand molecules and standard ligand molecules. 22
Results
PubChem screening of potential compounds
With standard ligand molecules apigenin and isorhapontigenin as the query molecules, a database of 1598 molecules was obtained as output. These molecules were further tested for Lipinski’s rule of five and among 1598 molecules, 24 compounds clearing Lipinski’s rule of five were further saved in Excel format for ADMET and druglikeness test.
ADMET and Druglikeness analysis of database
Among 24 molecules, 07 compounds cleared both ADMET and druglikeness tests when tested in ADMETlab software (Table 1 and Table 2). Molecules clearing both ADMET and druglikeness test were further selected with the help of Python script as the ligand molecules to test their docking affinity with the target protein hemagglutinin through AutoDock vina package (PyRx).
Table 1: Druglikeness results for tested compounds
Sl No |
Compound Name | MW | HBA | HBD | TPSA | No Rotatable bonds | RO5 violation |
| 1. | 5,7,4’-Trihydroxy-3-methoxyflavanone | 272.07 | 5 | 3 | 86.99 | 1 | 0 |
| 2. | 5,7,4′-Trihydroxyflavone | 360.12 | 7 | 1 | 83.45 | 5 | 0 |
| 3. | Catechin | 288.06 | 6 | 4 | 107.22 | 1 | 0 |
| 4. | 5-Hydroxy-7,3′,4′-trimethoxyflavone | 272.07 | 5 | 3 | 86.99 | 1 | 0 |
| 5. | Coumarin | 288.1 | 5 | 3 | 86.99 | 5 | 0 |
| 6. | Genistein | 220.07 | 4 | 1 | 59.67 | 1 | 0 |
| 7. | 3′,5-Dihydroxy-7,4′-dimethoxyflavone | 304.06 | 7 | 5 | 127.45 | 1 | 0 |
| 8. | Dihydroflavonol | 272.07 | 5 | 3 | 86.99 | 1 | 0 |
| 9. | 5,7-Dihydroxy-4′-methoxyflavone | 220.07 | 4 | 1 | 59.67 | 1 | 0 |
| 10. | Kaempferol | 288.06 | 6 | 4 | 107.22 | 1 | 0 |
| 11. | Epicatechin | 288.06 | 6 | 4 | 107.22 | 1 | 0 |
| 12. | Chlorogenic Acid | 224.07 | 5 | 2 | 75.99 | 4 | 0 |
| 13. | 2-Methoxy-4-(3-methyl-2-butenyl)phenol | 212.1 | 4 | 2 | 58.92 | 5 | 0 |
| 14. | Myricetin 3′,4′,5′-Tri-O-methyl Ether | 306.11 | 6 | 2 | 77.38 | 5 | 0 |
| 15. | Chrysin | 316.13 | 5 | 1 | 57.15 | 6 | 0 |
| 16. | Caffeic Acid | 212.07 | 5 | 3 | 86.99 | 4 | 0 |
| 17. | Resveratrol | 374.17 | 6 | 2 | 77.38 | 8 | 0 |
| 18. | 5,7,4′-Tri-O-methylkaempferol | 390.2 | 6 | 2 | 77.38 | 9 | 0 |
| 19. | 3,5,7,3′,4′-Pentamethoxyflavone | 350.14 | 7 | 2 | 86.61 | 8 | 0 |
| 20. | 7-Hydroxy-3′,4′-dimethoxyflavone | 290.12 | 5 | 1 | 57.15 | 5 | 0 |
| 21. | Vanillyl Alcohol | 318.15 | 5 | 2 | 68.15 | 7 | 0 |
| 22. | Chrysin | 316.13 | 5 | 1 | 57.15 | 6 | 0 |
| 23. | Methyl Syringate | 290.08 | 6 | 4 | 107.22 | 4 | 0 |
| 24. | Chlorogenic Acid | 224.07 | 5 | 2 | 75.99 | 4 | 0 |
Table 2: ADMET results for tested compounds
| Sl No | Compound CID | Absorption | Distribution | Metabolism | Elimination | ||||||
| Pgp-inh | Pgp-sub | Caco-2 | BBB | PPB | VDss | CYP-inh | CYP-sub | CL | T12 | ||
| 1. | 5,7,4’-Trihydroxy-3-methoxyflavanone | 0.003 | 0.023 | -5.03 | 0.04 | 81.76 | 1.214 | 0.066 | 0.118 | 6.291 | 0.669 |
| 2. | 5,7,4′-Trihydroxyflavone | 0.146 | 0.001 | -4.737 | 0.03 | 79.13 | 0.679 | 0.469 | 0.94 | 10.099 | 0.266 |
| 3. | Catechin | 0.01 | 0.024 | -5.095 | 0.067 | 86.57 | 0.905 | 0.571 | 0.309 | 14.619 | 0.776 |
| 4. | 5-Hydroxy-7,3′,4′-trimethoxyflavone | 0.002 | 0.011 | -5.193 | 0.067 | 82.08 | 1.686 | 0.107 | 0.107 | 4.65 | 0.68 |
| 5. | Coumarin | 0.029 | 0.01 | -4.77 | 0.043 | 89.72 | 0.601 | 0.957 | 0.854 | 14.77 | 0.906 |
| 6. | Genistein | 0.001 | 0.233 | -4.726 | 0.06 | 85.17 | 0.819 | 0.964 | 0.97 | 7.703 | 0.501 |
| 7. | 3′,5-Dihydroxy-7,4′-dimethoxyflavone | 0.007 | 0.023 | -6.08 | 0.036 | 84.06 | 1.139 | 0.059 | 0.105 | 8.411 | 0.822 |
| 8. | Dihydroflavonol | 0.004 | 0.01 | -4.778 | 0.185 | 82.08 | 1.758 | 0.794 | 0.457 | 14.899 | 0.592 |
| 9. | 5,7-Dihydroxy-4′-methoxyflavone | 0 | 0.268 | -4.716 | 0.043 | 89.98 | 0.751 | 0.98 | 0.96 | 3.539 | 0.507 |
| 10. | Kaempferol | 0.004 | 0.008 | -5.648 | 0.033 | 87.55 | 1.17 | 0.083 | 0.105 | 5.989 | 0.753 |
| 11. | Epicatechin | 0.006 | 0.009 | -5.143 | 0.089 | 60.55 | 0.752 | 0.15 | 0.101 | 12.089 | 0.765 |
| 12. | Chlorogenic Acid | 0.003 | 0.023 | -5.03 | 0.04 | 81.76 | 1.214 | 0.066 | 0.118 | 6.291 | 0.669 |
| 13. | 2-Methoxy-4-(3-methyl-2-butenyl)phenol | 0.021 | 0.05 | -4.797 | 0.211 | 86.08 | 0.672 | 0.131 | 0.946 | 11.702 | 0.814 |
| 14. | Myricetin 3′,4′,5′-Tri-O-methyl Ether | 0.001 | 0.124 | -4.439 | 0.249 | 33.49 | 1.22 | 0.83 | 0.847 | 11.245 | 0.895 |
| 15. | Chrysin | 0.091 | 0.155 | -4.843 | 0.027 | 87.17 | 0.547 | 0.671 | 0.968 | 11.632 | 0.896 |
| 16. | Caffeic Acid | 0.024 | 0.008 | -4.855 | 0.169 | 87.72 | 0.919 | 0.751 | 0.971 | 9.411 | 0.817 |
| 17. | Resveratrol | 0.001 | 0.253 | -5.269 | 0.068 | 74.94 | 0.339 | 0.037 | 0.492 | 14.877 | 0.939 |
| 18. | 5,7,4′-Tri-O-methylkaempferol | 0.221 | 0.274 | -4.819 | 0.177 | 88.82 | 0.624 | 0.24 | 0.952 | 11.158 | 0.824 |
| 19. | 3,5,7,3′,4′-Pentamethoxyflavone | 0.205 | 0.175 | -4.757 | 0.058 | 85.6 | 0.889 | 0.231 | 0.955 | 13.105 | 0.732 |
| 20. | 7-Hydroxy-3′,4′-dimethoxyflavone | 0.003 | 0.008 | -4.555 | 0.234 | 70.08 | 2.287 | 0.439 | 0.347 | 10.972 | 0.833 |
| 21. | Vanillyl Alcohol | 0.212 | 0.106 | -4.649 | 0.078 | 87.75 | 0.552 | 0.914 | 0.96 | 10.781 | 0.802 |
| 22. | Chrysin | 0.046 | 0.052 | -4.77 | 0.27 | 81.49 | 0.724 | 0.217 | 0.947 | 12.106 | 0.863 |
| 23. | Methyl Syringate | 0.017 | 0.059 | -4.779 | 0.204 | 85.83 | 0.69 | 0.174 | 0.948 | 11.449 | 0.838 |
| 24. | Chlorogenic Acid | 0.005 | 0.004 | -5.135 | 0.006 | 81.8 | 0.73 | 0.6 | 0.673 | 18.544 | 0.891 |
Target protein preparation in UCSF Chimera
The target protein selected for molecular docking was prepared in the UCSF chimera using the AutoDock
package. The grid coordinates surrounding crystallized ligands were used as input for site-specific docking. Figure 1 indicates the removal of miscellaneous molecules and the addition of charge and hydrogen to target protein to facilitate the molecular docking process.
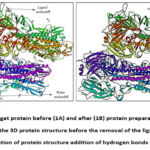 |
Figure 1: Target protein before (1A) and after (1B) protein preparation process. |
Ligand preparation for molecular docking
Ligand molecules were selected for molecular docking and were prepared using Open Babel software and PyRx AutoDock vina package. Figure 2 indicates the energy minimization process performed to stabilize the ligand molecules for the molecular docking process.
 |
Figure 2: Energy minimization process used for preparation of ligand structure for molecular docking process depicted using standard ligand as an example. |
Molecular docking of ligands with hemagglutinin
Among 07 ligand molecules, ligand molecule 5,7,4’-Trihydroxy-3-methoxyflavanone (PubChem ID 72281)25 displayed the highest docking score of -8.5 kcal/mol, lowest RMSD of 0.00 A0 in comparison with the standard drug Apigenin, which displayed a docking score of -8.4 kcal/mol (Table 3). Hence the ligand compound 5,7,4’-Trihydroxy-3-methoxyflavanone can be further selected for protein-ligand interaction analysis.
Table 3: Molecular docking results for tested compounds
| S. No | Name of Compound | Score (Kcal/mol) | Interaction | PubChem ID |
| 1. | 5-Hydroxy-7,3′,4′-trimethoxyflavone | -8.0 | 04 | 5272653 |
| 2. | 3′,5-Dihydroxy-7,4′-dimethoxyflavone | -8.4 | 06 | 5378823 |
| 3. | 5,7,3′,4′-Tetramethoxyflavone | -8.3 | 06 | 631170 |
| 4. | 5,7,4’-Trihydroxy-3-methoxyflavanone | -8.5 | 06 | 72281 |
| 5. | 3,5,7,3′,4′-Pentamethoxyflavone | -8.4 | 04 | 97332 |
| 6. | 5,7-Dihydroxy-4′-methoxyflavone | -7.3 | 03 | 5280442 |
| 7. | 7-Hydroxy-3′,4′-dimethoxyflavone | -7.4 | 04 | 5378518 |
| Apigenin (Standard) | -8.4 | 06 | 5280443 |
Protein-ligand analysis in Discovery Studio
Protein-ligand interaction analysis depicted that the molecule 5,7,4’-Trihydroxy-3-methoxyflavanone and standard drug apigenin formed 06 bonds with the active site amino acids present inside the active site of the target protein hemagglutinin. Figure 3 indicates a 3D protein-ligand interaction analysis of protein hemagglutinin with molecules 5,7,4’-Trihydroxy-3-methoxyflavanone and apigenin. Figure 4 indicates the 2D interaction between both ligand molecules (5,7,4’-Trihydroxy-3-methoxyflavanone and apigenin) with the active site amino acids of the target protein hemagglutinin. Hence ligand molecule 5,7,4’-Trihydroxy-3-methoxyflavanone isolated from the plant Populus szechuanica can be further developed as a novel effective drug against the target protein hemagglutinin to treat influenza.
 |
Figure 3: 3D protein-ligand interaction analysis of protein hemagglutinin with molecules 5,7,4’-Trihydroxy-3-methoxyflavanone (Red) and standard drug apigenin (Dark blue). |
 |
Figure 4: Protein-ligand interaction analysis between ligand molecules and target protein hemagglutinin. |
Discussion
The influenza virus, a global health threat, is responsible for many respiratory deaths and cardiovascular deaths annually. The present antiviral drugs are unable to tackle the virus completely due to its rapid mutational rates. Hence there is a need to find novel and effective antiviral drugs to address the influenza viral infection and reduce the death rates significantly. Central to the influenza virus’s life cycle is the hemagglutinin protein, the primary target for antiviral drug development. Among the potential target proteins studied for the influenza virus, the target protein hemagglutinin plays a vital role by allowing the entry of the virus inside the human body.26 Hence the present study aims to identify potent inhibitors of hemagglutinin by focusing on the pharmacological potential of similar compounds to the known compounds apigenin and isorhapontigenin. The presented study screened a library of molecules retrieved from PubChem database, rigorously testing them against Lipinski’s rule of five, ADMET properties, and drug-likeness criteria. From an initial pool of 24 molecules, 07 candidates cleared both Lipinski’s rule of five and the ADMET test and were further selected as ligand molecules for the molecular docking process against the target protein hemagglutinin. Among the 07 tested compounds, phytocompound 5,7,4’-Trihydroxy-3-methoxyflavanone (PubChem ID 72281),25 isolated from plant Populus szechuanica and retrieved from PubChem database emerged as the leading candidate, exhibiting the highest docking score of -8.5 kcal/mol and formed 06 bonds with the 06 active site amino acids of the target protein hemagglutinin. Sadati and the team have studied the flavonoids through molecular docking to find novel inhibitors for the target protein hemagglutinin against influenza disease. Results indicated that the best compound indicated in the study displayed a docking score of -7.5 kcal/mol, while on the other hand, the phytocompound 5,7,4’-Trihydroxy-3-methoxyflavanone discussed in our study displayed a docking score of -8.5 kcal/mol and formed 06 bonds with the 06 active site amino acids of the target protein hemagglutinin. Further, our study has performed druglikeness and ADMET study to evaluate the safety and toxicity of phytocompound 5,7,4’-Trihydroxy-3-methoxyflavanone inside the human body, which is missing in the study conducted by Seyed and his team.23 Chinayan and his team have performed a study to explore novel compounds to block the activity of haemagglutinin protein as a medication against influenza virus. The study only focused on molecular docking analysis, while on the other hand, our study has explored druglikeness and ADMET study to evaluate the safety and toxicity of phytocompound 5,7,4’-Trihydroxy-3-methoxyflavanone inside the human body, which is missing in the study conducted by Seyed and his team.24 The compound homoeriodictyol has been isolated by Okinczyc and his team from the bud exudates of Populus szechuanica and stored in the PubChem database. A study analyzing the phenolic compounds in the bud exudates of Populus szechuanica.27 These findings indicated that the phytocompound 5,7,4’-Trihydroxy-3-methoxyflavanone can be further explored as a novel and effective lead compound against influenza virus with hemagglutinin as the target protein through in-vitro and in-vivo tests.
Conclusion
The flu, caused by the influenza virus is one of the leading causes of death around the world. The present medications are unable to fully address the virus due to the rapid mutation of the virus. In the present study, the ligand molecule 5,7,4’-Trihydroxy-3-methoxyflavanone (PubChem ID 72281) shows a docking score of -8.5 kcal/mol and zero RMSD value. Hence, the molecule 5,7,4’-Trihydroxy-3-methoxyflavanone can be further tested in vitro and developed into a suitable drug for influenza virus.
Acknowledgment
The authors acknowledge support from REVA University for the facilities provided to conduct the present research work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
DATA AVAILABILITY:
This statement does not apply to this article
Data Availability Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to Produce Material From Other Sources
Not Applicable
Author Contributions
- Raje Siddiraju Upendra: Conceptualization and Original Draft.
- Mattur Srinivasa Murthy Upamanyu: Data Collection and Analysis
- Sanjay Shrinivas Nagar: Methodology and Writing Original Draft
- Preetham Rechihalli Shivalingappa and Sandeep Anjaneya: Data Visualization and Resources
- Karthik Rajendra: Supervision and Project Administration.
References
- European Centre for Disease Prevention and Control. Avian Influenza Overview March – June 2024. ECDC. Jul. 12, 2024. [Online]. Available: https://www.ecdc.europa.eu/en/publications-data/avian-influenza-overview-march-june-2024#:~:text=Between%2013%20March% 20and%2020,and%20Me %20Mexico%20(one%20fatal%20A. [Accessed: Aug. 19, 2024].
- Cao N, Cai Y, Huang X, Jiang H, Huang Z, Xing L, Lu L, Jiang S, Xu W. Inhibition of influenza A virus and SARS-CoV-2 infection or co-infection by griffithsin and griffithsin-based bivalent entry inhibitor. mBio. 2024;15(2):e00741-24.
CrossRef - Tosheva II, Saygan KS, Mijnhardt SM, Russell CJ, Fraaij PL, Herfst S. Hemagglutinin stability as a key determinant of influenza A virus transmission via air. Opin. Virol. 2023;61:101335.
CrossRef - Lakshmi SJ, Siddiraju UR. A comprehensive study of secondary metabolite profile and pharmacological effects of medicinal plant Toddalia asiatica. Appl. Pharm. Sci. 2022;12(7):042–052.
CrossRef - Priyanka, R., Reddy, A. H., Manokaran, S., & Siddiraju, U. R. Antiproliferative effects of aromatica Extracts and Essential oils in MDA-MB231 as a Breast Cancer Cell Line. RJPT. 2024; 17(3): 1352-1355.
CrossRef - Cho WK, Choi HJ, Ma JY. Selaginella tamariscina ethanol extract attenuates influenza A virus infection by inhibiting hemagglutinin and neuraminidase. Nutrients. 2024;16(14):2377
CrossRef - National Center for Biotechnology Information (2025). PubChem Compound Summary for CID 5280443, Apigenin. Retrieved January 3, 2025, from https://pubchem.ncbi.nlm.nih.gov/compound/Apigenin.
- National Center for Biotechnology Information (2025). PubChem Compound Summary for CID 5318650, Isorhapontigenin. Retrieved January 3, 2025, from https://pubchem.ncbi.nlm.nih.gov/ compound/ Isorhapontigenin.
- Rolta R, Salaria D, Fadare OA, Fadare RY, Masih GD, Prakash A, Medhi B. Identification of novel inhibitor phytoconstituents for Influenza A H3N2: an in-silico J Biomol Struct Dyn. 2024;42(8):4287–4296.
CrossRef - Haritha, M., Sreerag, M., & Suresh, C. H. Quantifying the hydrogen-bond propensity of drugs and its relationship with Lipinski’s rule of five. NJC, 2024; 48(11): 4896-4908.
CrossRef - Ramos DBM, Araújo MTMF, Araújo TCL, Napoleão TH. Evaluation of antitumor activity and toxicity of Schinus terebinthifolia leaf extract and lectin (SteLL) in sarcoma 180-bearing mice. J Ethnopharmacol. 2019;235:148–157
CrossRef - Upendra RS, Nagar SS, Vasudevan K. Computational screening of phytocompounds isolated from the plant Toddalia asiatica and Coleonema album as potential inhibitors against enzyme DPP-4 for Type-2 diabetes mellitus treatment. Chemical Papers. 2024;78(3):1833–1847.
CrossRef - Shinde SS, Giram PS, Wakte PS, Bhusari SS. ADMET tools in the digital era: Applications and limitations. Pharmacol. 2025;91:1–25
CrossRef - Fu L, Shi S, Yi J, Wang N, He Y, Wu Z, Cao D. ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality, and decision support. Nucleic Acids Res. 2024;52:gkae236.
CrossRef - Trovão, N. S., Khan, S. M., Lemey, P., Nelson, M. I., & Cherry, J. L. Comparative evolution of influenza A virus H1 and H3 head and stalk domains across host species. Mbio. 2024; 15(1): e02649-23.
CrossRef - Siddiraju UR, Rajendra K, Nagar S, Prasad H, Chakki K, Kulkarni VP, Preetham. Computational identification of novel Leukotriene A4 Hydrolase (LTA4H) inhibitors as therapeutic candidates for colorectal cancer. Integr. Sci. Technol. 2024;12(1):45–56
- Priyanka, P., & Anbarasu, K. In-silico analysis on human FTO and mutant S319F with various ligands for poly malformation syndrome. AIP Conf. Proc. 2024; 3193(1).
CrossRef - Ozgul M, Katz B, Nashine S, Kenney MC. Stability determination of HNF14 in H2O and PBS using high-resolution mass spectrometry. IOVS. 2023; 64(8):3014.
- Stankunas E, Köhler A. Docking a flexible basket onto the core of the nuclear pore complex. Nat Cell Biol. 2024;26(8):1504–1519
CrossRef - Tahir M, Baharuddin M, Najib A. In silico screening of brotowali (Tinospora crispa) chemical compounds as α-glucosidase inhibitor using the PyRx program. AIP Conf. Proc. 2023; 2595(1).
CrossRef - Sharma, S., Sharma, A., & Gupta, U. Molecular Docking studies on the Anti-fungal activity of Allium sativum (Garlic) against Mucormycosis (black fungus) by BIOVIA discovery studio visualizer 21.1. 0.0. JAA. 2021; 5(1): 028-032.
CrossRef - Begum A, Ameen, Nagar SS, Upendra RS, Karthik R. Discovery of novel antifungal agents from Polygonatum odoratum targeting 14-alpha demethylase enzyme for the treatment of black fungus (mucormycosis). Presented at: 2024 IEEE International Conference on Intelligent Systems and Advanced Applications (ICISAA); 2024 Oct.
CrossRef - Sadati, S. M., Gheibi, N., Ranjbar, S., & Hashemzadeh, M. S. Docking study of flavonoid derivatives as potent inhibitors of influenza H1N1 virus neuraminidase. Biomedical reports. 2019;10(1): 33-38.
CrossRef - Indumathi, C. P., Bupesh, G., Vasanth, S., Senthilkumar, V., Anandh, A. V., & Pandian, K. Molecular docking analysis of zanamavir with haem agglutinin neuraminidase of human para influenza virus type 3. Bioinformation. 2019;15(10): 730.
CrossRef - National Center for Biotechnology Information. PubChem Compound Summary for CID 73635, Homoeriodictyol. NCBI. Apr. 18, 2025. [Online]. Available: https://pubchem.ncbi.nlm.nih.gov/compound/ Homoeriodictyol [Accessed: Apr. 18, 2025].
- Lee J, Kim Y, Lee Y, Lee J, Kim H, Kim Y. Antiviral effect of isoquercitrin against influenza A viral infection via modulating hemagglutinin and neuraminidase. Int J Mol Sci. 2022;23(21):13112.
CrossRef - Okińczyc P, Widelski J, Nowak K, Radwan S, Włodarczyk M, Kuś PM, Susniak K, Korona-Głowniak I. Phytochemical profiles and antimicrobial activity of selected Populus spp. bud extracts. Molecules. 2024;29(2):437.
CrossRef
Abbreviations
HA: Hemagglutinin
ADMET: Adsorption, Distribution, Metabolism, Excretion and Toxicity
PDB: Protein Data Bank
PDBQT: Protein Data Bank Partial Charge Atom Type
BBB: Blood Brain Barrier
QED: Quantum Electrodynamics

