
Seruni Kusuma Udyaningsih Freisleben1 and Hans-Joachim Freisleben2*
and Hans-Joachim Freisleben2*
1Department of Physics, Faculty of Mathematics and Natural Sciences, Universitas Indonesia, Campus Depok, Depok, Indonesia
2Medical Research Unit, Faculty of Medicine, Universitas Indonesia, Campus Salemba, Salemba Raya, Jakarta, Indonesia
Corresponding Author; E-mail address: hj.freisleben@t-online.de
DOI : https://dx.doi.org/10.13005/bpj/3037
Abstract
Thalassemic hemoglobin (Hb) differs from normal Hb in its structure and oxygen-binding capacity. To find out structural differences in their heme-iron sites, X-ray absorption fine structure (XAFS) was applied. Resulting data in the range of 0 ≤ k ≤1 6 Å-1 and 1 ≤ k ≤ 14 Å-1 obtained from frozen aqueous solutions (10 K) of normal and thalassemic HbE/F deoxy forms were refined using multiple-scattering (MS) analyses. To test the robustness of the refinements R starting models, constraints, restraints, and k ranges were varied; for normal relaxed R- and tense T-deoxy-Hb final XAFS R values were 13% and 17% and for thalassemic HbE/F relaxed R-deoxy-Hb R value was 19-21%. With normal R-deoxy-Hb, we obtained Fe-Np (pyrrole) and Fe-N€(imidazole) distances of 2.05 Å and 2.14 Å, whereas in thalassemic HbE/F R-deoxy-Hb, these values were 2.11Å and 1.9Å , respectively. In the T-state of normal deoxy-Hb, Fe-Np distance (2.08 Å) was longer and Fe-N€ (1.96 Å) shorter than in the R-state; from thalassemic HbE/F T-deoxy-Hb we could not obtain sufficiently stable probes for XAFS measurement. In normal and HbE/F R-deoxy-Hb the Fe-His imidazole bond deviated from the z-axis by ~100, whereas it was on the z-axis in the T-state of normal Hb. Heme doming increased from normal R-deoxy via normal T-deoxy to thalassemic R-deoxy forms with simultaneously increasing dislocation of the central iron from the plane of the porphyrin. Oxygenation brings the iron back into the plane of the porphyrin ring.
Keywords
Beta-thalassemia; Heme fine Structure; Hemoglobin; Synchrotron; Thalassemic HbE/F; X-ray absorption fine structure (XAFS)
Download this article as:| Copy the following to cite this article: Freisleben S. K. U, Freisleben H. J. X-ray Analysis of Fine Structure of Heme in Normal and Thalassemic HbE/F Hemoglobin. Biomed Pharmacol J 2024;17(4). |
| Copy the following to cite this URL: Freisleben S. K. U, Freisleben H. J. X-ray Analysis of Fine Structure of Heme in Normal and Thalassemic HbE/F Hemoglobin. Biomed Pharmacol J 2024;17(4). Available from: https://bit.ly/4gQLN34 |
Introduction
Heme proteins participate in a wide range of enzymatic redox processes, in electron transfer, and in the transportation and storage of dioxygen. Hemoglobin (Hb) of normal adults is 95% HbA with two alpha and two beta globin chains (α2β2). Minor Hb species are HbA2 consisting of two alpha and two delta globins (a2dδ2), which amounts to 2-3.5% of total Hb and fetal Hb (HbF) made up of two alpha and two gamma globins (a2ϒ2), which is less than 2%. At the third semester of pregnancy, HbF (up to 90% of Hb in maternal blood) synthesis starts to decline and is gradually replaced by adult HbA (α2β2) over the first year after birth.1-3
Thalassemia is a hereditary Hb disease characterized by the absence or decreased production of normal globin chains which leads to varying degrees of anemia.4-6 In Indonesia, a G to A shift in codon 26 causes the most common (beta-26 Glu to Lys) mutation (HbE) frequently associated with beta-thalassemia.7-11 Splice site mutation in exon 1 of the beta-globin chain makes HbE abnormal leading to microcytic anemia11 and the clinical picture of thalassemia intermedia, especially when combined with further beta-thalassemic mutations or deletions. In thalassemia patients very often the production of fetal Hb does not drop off at birth but is maintained because it is still necessary for their survival.2
On chromosome 16 are the genes which control ɑ-globin production and on chromosome 11 the genes are located for β, γ, and δ globin synthesis. The β-globin chain which is made up of 146 amino acids binds heme covalently via the proximal histidine in position 92 (His-92) and alterations in the structure of the β-globin chain in hereditary Hb diseases mostly lead to an overproduction of HbF.1 A distal His, in position 63, is not directly involved heme binding,12 but may function as a ligand to the central Fe. Thus, the heme is fixed between helices E and F of the globin chain. Several amino acid residues form the hydrophobic heme pocket around the protoporphyrin and the two histidines. With distances less than 6 Ǻ from the heme, Leu-28, Leu-31, Thr-38, Phe-41, Phe-42, Phe-45, Val-67, Leu-68, Ala-70, Phe-71, Phe-85, Leu-88, Leu-91, Leu-96, Val-98, Asn-102, Phe-103, Leu-106, Val-137, Leu-141 provide hydrophobic interaction, whereas the phenylalanines (Phe) may, in addition, interact via aromatic contact, and (besides the two 63 and 92 histidines) only two amino acid residues, Ser-44 and Lys-66, form hydrogen bonds within the heme pocket.13
Various spectroscopic methods, such as ultraviolet (UV) and visible spectroscopy, infrared, resonance Raman (RR), microwave spectroscopy and X-ray crystallography have been used to investigate heme proteins such as met-indoleamine 2,3-dioxygenase-2,14 myoglobin,15-17 and Hb.2,11,18-20 Whereas structural changes in thalassemic globin chains have been widely reported, fine structure of their heme has not been known. Here we investigate the heme structure of a patient with 2/3 HbE and 1/3 HbF.
Materials and Methods
To better understand the chemistry and dynamics of Hb, model compounds have been constructed, such as the “picket fence” complex21 or meso-(2-methylimidazole)[tetraphenyl-porphyrinato(2-)]iron(II).22 In both structures modeling the oxygenated heme complex, the bonds from iron to the porphyrin nitrogens are 0.09 Å shorter than in the deoxygenated complexes and the bond from the iron to the imidazole nitrogen deviates by 10.3o from the z-axis. Such models served as references for our study.
Sample Preparation
Blood from a 13-year-old non-transfused thalassemia intermedia patient with 1/3 HbF and 2/3 HbE was used for XAFS analysis. The reasons for this decision were: i) this non-transfused patient did not have mixtures of endogenous and exogenous Hb in his blood (as transfused patients do); ii) in Hb analysis, the patient had a clear relation of 67.5% HbE and 32.5% HbF; iii) frequently, patients show more complex Hb composition (including varying contents of thalassemic Hb and/or HbA2), which complicates interpretation of XAFS data; iv) this patient had already been included in comprehensive thalassemia examinations on iron status, oxidative stress, and electron paramagnetic resonance (EPR) measurements of his isolated erythrocyte membrane.23-25 Hence, the chosen sample may also serve as a model for the wide variety of thalassemic hemoglobins.
Blood samples were drawn from patients of the thalassemia ward in Cipto Mangunkusumo General Hospital in Jakarta after fully informed consent which conforms to the standards applied in Indonesia. The study was approved by the Ethical Clearance Committee of the Faculty of Medicine, University of Indonesia and hemolysate samples were transported on dry ice from Jakarta to Sydney, Australia (AQIS permit No 200012081). The Hb content was 0.5–2 mM, as measured by UV/vis spectroscopy (a Hewlett-Packard 8452-A diode array spectrophotometer or a CARY 5E spectrophotometer) at 406 nm (met-Hb) against a standard prepared from commercial met-Hb (Sigma Australia). The literature value of the molar extinction coefficients ε (406 nm) = 162 M-1 cm-1 was used to determine concentration.26
Control Hb A was purchased from Sigma Australia in the form of met-Hb. The molecular or formula weight (FW) was given as 64,000, equivalent to four heme moieties of FW 16,000, each (including the respective alpha- or beta-peptide chains). For XAFS experiments, 250 μL of a solution containing 2 mM met-Hb (equivalent to 8 mM heme) was prepared with 60% bis-Tris (0.05 M) and 40% glycerol (v/v).15,16 The glycerol was added to reduce random noise scattering from the sample surface and to prevent potential damage to the protein from ice crystals as well as interference with Bragg reflections from ice crystals in the XAFS spectra.
Control deoxy-Hb was prepared by reduction of met-Hb with sodium dithionite (6- to 10-fold molar concentration vs. heme in bis-Tris buffer 0.05 M, pH 6.5) using syringe techniques in an argon-filled glove bag.15,16
HbE/F solution (500 μL, 0.5-2.0 mM) was oxidized with K3[Fe(CN)6] (0.003 mg, 0.02 mM) and the solution was purified by passing it down a column of Sephadex G25-p10. The pooled red fractions were concentrated by dialysis against PEG400. The final met-Hb concentration for XAFS experiments was determined from UV/Vis spectroscopy to be 2.0-2.6 mM, equivalent to 8.0-10.4 mM heme, in NaPi buffer (5 mM, pH 6.8). After hemolysis and oxidation, met-HbE/F was already dissolved in NaPi buffer (5 mM, pH 6.8). Hence, reduction of HbE/F to deoxy-HbE/F was also carried out in this buffer (NaPi, 5 mM, pH 6.8), but it needed a higher concentration of sodium dithionite (at least 10-fold over heme concentration) to reduce met-HbE/F.
All handling of proteins was undertaken using protein-clean glassware and every step in the Hb preparation was checked by UV/vis spectroscopy at wavelengths between 200 and 700 nm.27
X-ray Absorption Fine Structure (XAFS)
Numerous scans were collected and signal-averaged for a 1-2 mM Hb sample, equivalent to 4-8 mM heme. Solutions of Hb (6-8 mM heme) were syringed into a 140-µL Lucite cell with Mylar tape windows in an Ar-filled glove bag and frozen in liquid N2 /n-hexane. A low temperature (10 K) was maintained using an Oxford Instruments continuous-flow liquid He CF 1208 Cryostat. The energy was calibrated using an iron (Fe foil) standard (first inflection point, 7111.2 eV). Iron K-edge X-ray absorption spectra (XAS) were recorded at the Stanford Synchrotron Radiation Laboratory (SSRL) on the beamlines 9-3 and 7-3 under dedicated ring conditions (3 GeV, 50-100 mA) using a Si (220) double-crystal monochromator.
We averaged the fluorescence XAS scans by rating the signal-to-noise ratio. The spectra obtained from each of the detector channels were checked and poor data due to noisy detector channels, low detector channel sensitivity, or those affected by step jumps, were removed from the averaged data because they resulted in a quality decrease of the averaged XAS and, hence, the accuracy of the XAFS fit. The background was corrected towards the pre-edge region and the fit was extrapolated into the XAFS region. Monochromator glitches were edited following calibration and subtraction of the background absorbance. For the removal of the underlying absorbance, a spline of three to five regions was fitted to the XAFS region and subtracted (Table 1). The resulting data were normalized to an edge jump of 1.0 and compensated for decreasing absorbance past the edge using a k3-weighting, so that the final XAFS was relative to the edge (μ0), and the amplitude enhanced at high values of the photoelectron wave vector k. After the background was subtracted, data were normalized, compensated for k3, and plotted as a function of k.28
For the fits, already published heme models were used including that of meso-(2-methylimidazole)[tetraphenyl-porphyrinato(2-)]iron(II) with the imidazole ring of the histidine side chain as a reference. The starting bond lengths and angles were taken from the Protein Data Bank, ID: IGZX (T-oxy-Hb) and IC7B (R-deoxy-Hb). The model-fitting calculations were performed using the XFIT program,28 which incorporates ab initio calculation of the XAFS using MS (version 6.01) of FEFF.
Table 1: Spline and Refinement Parameters
| Hemoglobin | Pre-edge Background (eV) | Spline Segments (eV) | S02 | R (%) |
| Normal-R-deoxy- | 7094.7 – 7098.7 | 7130 – 7211.37211.3 – 7414.27414.2 – 7823.87823.8 – 8257.4 | 0.98 | 13 |
| Normal-T-deoxy- | 7072.35 – 7087.11 | 7134.2 – 7355.37355.3 – 7821.47821.4 – 8244.3 | 1.1 | 17 |
| Thalassemic-Hb E/F | 7075.2 –7108.8 | 7135.0 – 7310.37310.3 – 7381.97381.9 – 7623.47623.4 – 7913.9
7913.9 – 8784.5 |
1.15 | 19-21 |
Table 1. Legend
Data were collected from samples containing 6-8 mM Fe on the beamlines 9-3 and 7-3 under ring conditions, 3 GeV, 50-100 mA, using a Si (220) double-crystal monochromator and 13- and 30-element Ge array detectors. The XAFS (k‑space) window parameters were varied in the range 1‑14 to 1-16 Å–1. The (r‑space) window parameters were 0‑5.0 and 0.75-5.0 Å for the multiple‑scattering analysis of normal deoxy-Hb and thalassemic Hb, respectively. The data were fitted according to previous work done with deoxy-leghemoglobin and deoxy-myoglobin at 10 K.15,16,29
Results
Photodecomposition Effects
To investigate any photodecomposition effects on the samples, the edge shifts from the first scan to the last scan were observed. Due to the negligible photodamage in the edge data of all Hb samples (the edge shifted less than 0.1 eV compared to the first scan), all scans were incorporated into the analyses. The edge positions of the T-forms were higher in energy than the R-forms.
X-Ray Absorption Fine Structure (XAFS)
The parameters of interest: bond lengths between the iron and the four protoporphyrin nitrogens (Fe-Np); the distance between the iron and the imidazole nitrogen of the proximal histidine (Fe-Nε); whether the iron is located in the plane of the protoporphyrin ring or how far it is located out of the plane; changes in these parameters with changes from R- to T-states of the Hb; the distance between the iron center and the dioxygen in oxy-Hb; and the respective bond angles.
Multiple-Scattering Refinement of the Iron Site
The observed and calculated XAFS, Χ(k) × k3, the corresponding Fourier transforms, the residuals, ∆[χ(k) × k3, and the window functions used in the Fourier filter for normal R and T deoxy-Hb and thalassemic deoxy-Hb are shown in Figure 1. The MS analysis indicates differences in the Fe–Ne of normal T-deoxy-Hb and of R-deoxy-Hb.
Distances of the Fe Atom from the Porphyrin Plane and the Imidazole Ring
The Fe–Np distance of the normal R-deoxy-Hb is 2.05 Å. The imidazole of the normal R-deoxy-Hb is closer to the heme plane (0.29 Å) than that in the T-form, which is located further away (0.4 Å). These values are consistent with the predicted changes based on X-ray crystallography. The thalassemic Fe-Np distance is longer (average 2.11 Å) than the distances for normal R- and T-deoxyhemoglobin (2.05 and 2.07 Ǻ, respectively). The porphyrin ring in the thalassemic R-deoxy-Hb looks distorted. However, in the R-oxy-Hb oxygenation brings the central iron back into the plane of the porphyrin ring system.
Table 2: XAFS Structures – Fe-Ligand Bond Length
| Hemoglobin | Fe-L bond length | Iron out of porphyrin plane | |
| Fe-Np | Fe-Nε | ||
| Normal R-deoxy | 2.05 Å | 2.13 Å | 0.29 Å |
| Normal T-deoxy | 2.07 Å | 1.95 Å | 0.40 Å |
| Thalassemic R-deoxy |
2.12 Å 2.10 Å |
1.87 Å 1.92 Å |
0.65 Å |
| Thalassemic R-oxy |
2.01Å | 2.0Å | – |
Table 2: Legend: Distances: Fe-L, Fe-ligand; Fe-Np, Fe-nitrogen(porphyrin); Fe-Nε, Fe-nitrogen(His-92 imidazole)
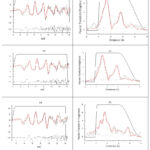 |
Figure 1: (a) XAFS curves and (b) Fourier transform; observed (black-dash), calculated (red) and residual (black-dot); of normal R-deoxy-Hb (upper panel), T-deoxy-Hb (middle panel), R-deoxy-HbE/F (bottom). |
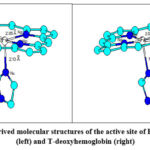 |
Figure 2: XAFS-derived molecular structures of the active site of R-deoxyhemoglobin (left) and T-deoxyhemoglobin (right) |
Structural differences between R- and T-deoxy-Hb are minute and are more evident from the values in Table 2 than from the structural models in Figure 2.
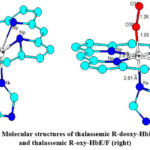 |
Figure 3: Molecular structures of thalassemic R-deoxy-HbE/F (left) and thalassemic R-oxy-HbE/F (right). |
The XAFS of thalassemic R-deoxy-HbE/F heme looks distorted with Fe located further out of the plane of the porphyrin ring system than in normal Hb reducing its oxygen affinity. Oxygenation in R-oxy-HbE/F brings the iron back to the plane of the porphyrin ring system (Figure 3).
Discussion
All dioxygen-binding hemeproteins have in common the prosthetic group, iron protoporphyrin IX, the heme group, located in a hydrophobic pocket of the protein. The heme is connected to the globin chain on the proximal side by a covalent bond between the iron atom and an imidazole-nitrogen (Nε) of the (proximal) histidine residue. Besides this covalent bond, the protoporphyrin is further fixed to the protein chain via hydrophobic forces. Only two hydrophilic residues are present on the heme moiety, i.e., propionates, which possibly interact electrostatically with external peptide residues, in an analogous way to the heme interactions in myoglobin.30 In all hemoglobins, the four nitrogens of the porphyrin (Np) and Nε of the proximal His coordinate the central iron: in deoxy-Hb, Fe is five-coordinate and paramagnetic with a spin of S = 2.22,31,32
In our measurements, the Fe–Np distance (2.05 Å) of the normal R-deoxy-Hb is 0.03 Å shorter compared to that of the deoxy-Hb investigated by Brucker,33 but similar to the Fe–Np distance (2.06 Å) in the myoglobin studied by Rich.15 The imidazole of the normal R-deoxy-Hb is closer to the heme plane (0.29 Å) than that in the T-form, which is located further away (0.4 Å). These values are consistent with the predicted changes based on X-ray crystallography. The thalassemic Fe-Np distance is much longer (average 2.11 Å) than those for normal R- and T-deoxy-Hb (2.05 and 2.07 Ǻ, respectively). The porphyrin ring in the thalassemic deoxy-Hb looks distorted. The extent of the distortion produced in the fit may be overrepresented which may, in part, be due to the mixture of HbF and HbE and the variation of the Fe-Np distance between 2.10 Å and 2.12 Å.
Normal R- and T-deoxy-Hb
The fact that the iron is out of the heme plane in the normal R-deoxy-Hb is consistent with the theory that the diameter of a high-spin t42ge2g Fe(II) ion is larger than that of the central hole in the porphyrin ring.32 In the oxygenated form, the complex converts to low spin (corresponding to the t42g configuration of Fe(II)), the Fe ion shrinks a little, and moves into plane.34
We do not want to go into the controversial views about the nature of R- and T-states of deoxy-Hb as discussed by Park and colleagues,35 however, we refer to their values of Fe deviation from the heme plane in 1.25 Å resolution crystal structures. The calculations of the authors differentiate between all 24 porphyrin atoms where the iron deviates from the plane by 0.2-0.3 Å and only the four porphyrin nitrogens, where the iron displacement from the plane is 0.3-0.4 Å. These values correlate well with our measurements of 0.29 Å (R-form) and 0.4 Å (T-form). In the T-form of normal deoxy-Hb, the iron is pulled out of the heme plane 0.11 Å further than in the R-form (0.29 Å) towards the proximal His, concomitantly it has lower affinity for dioxygen.36 The shift from R- to T-quaternary structure is consistent with the bond lengths of Fe-Np and Fe-Ne and the α1β2 contacts in crystal structures.37
Thalassemic deoxy-Hb and oxy-Hb
The iron in thalassemic R-deoxy-Hb moves even further towards the proximal histidine (0.65 Å) than in the T-form of normal deoxy-Hb. The T-form has lower oxygen affinity than the R-form,36 accordingly, oxygen affinity of thalassemic R-deoxy-Hb is even much lower. In analogy to normal deoxy-Hb, the iron in the thalassemic T-form should be displaced even further from the plane of the porphyrin. Apparently, the thalassemic Hb does not only exert lower oxygen affinity but also lower molecular stability than normal deoxy-Hb. We managed to produce sufficiently stable probes for XAFS measurements from normal deoxy-Hb both in the R- and T-forms, but only from the R-form of thalassemic deoxy-Hb. X-ray crystallography with normal Hb and HbS indicated that hydrophobic amino acids Phe-b85 and Leu-β85 stabilize the globin structure, Hb assembly, and heme binding.38 HbS variants containing Glu in one of these positions, or in both, destabilize the pocket structure, but it is uncertain as how this relates to the thalassemic mutants. Besides genetically caused structural destabilization39 oxidative stress by iron overload25 induces radical formation and additional destabilization of thalassemic hemoglobins.40
In their study, Rees and colleagues41 suggest that HbE itself is not unstable at normal body temperature, but may become unstable under oxidative stress at increased, febrile temperatures. Oxidative stress leads to the formation of free radicals within the globin chains including higher oxidative iron states and initiates a pathophysiological cascade of hemoglobin destabilization and damage of the erythrocyte membrane.42,43 HbE itself presents a rather mild clinical picture but the combination HbE/ß-thal can cause severe problems.44 In HbE/ß-thalassemia two thalassemia alleles interact in varying degree of globin chain imbalance, from which the marked variability in severity between different individuals with HbE/ß-thalassemia may principally result. The thalassemic Hb in our study contains 33% of HbF. Hirsch and colleagues42 stated that high levels of HbF are associated with more moderate clinical symptoms. In Hb E/ß-thal HbF levels have been reported to vary from 10% to 80%.45
Strader and his working group46 conclude that the occurrence of free ɑ-subunits increases the oxidative instability of HbE. Samuel and colleagues47 investigated the mechanism and intermediates of the disassembly and unfolding of hemoglobins which pathway may further contribute to instability.
Conclusion
On the one hand, we managed to show the X-ray absorption fine structures of thalassemic R-deoxy-HbE/F and R-oxy-HbE/F. On the other hand, our probes from T-deoxy-HbE/F were not sufficiently stable for XAFS measurements. We used one form of pathological Hb with a defined composition of 2/3 HbE and 1/3 HbF, exemplary and representative of thalassemic hemoglobins. However, there are several hundreds of mutations and deletions both in ɑ- or ß-thalassemias. Each of these mutations is likely to have its unique structural modification of the heme pocket of their respective Hb. There are also various combinations with HbE, for example, a rare case of Hb Constant Spring and HbE was recently reported from China.48 Hence, our study is a start and further investigations should be done with other forms of thalassemic Hb for comparison and to obtain a more comprehensive knowledge of thalassemic heme fine structure.
Acknowledgements
The authors express their deep thankfulness to the University of Sydney, especially to Professor Peter Lay, late Professor Robert Armstrong, and Dr. Aviva Levina for their support and guidance. Gratitude is acknowledged to the UC Stanford Synchrotron for providing the opportunity to measure XAFS of hemoglobins. Furthermore, HJF is grateful to the German Academic Exchange Service (DAAD) for his long-term professorship at the Medical Faculty of Universitas Indonesia.
Funding Sources
This study was funded as part of the URGE Project of the Study Program Biomedical Sciences at the Medical Faculty Universitas Indonesia with the title “Iron Overload and Oxidative Stress in Thalassemia Patients in Jakarta” and the No. 002/ PS/III/URGE/99.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
Additional data can be provided on request.
Ethics Statement
The study was approved by the Ethical Clearance Committee of the Faculty of Medicine, University of Indonesia and hemolysate samples were transported on dry ice from Jakarta to Sydney, Australia (AQIS permit No 200012081).
Informed Consent Statement
Blood samples were drawn from patients of the thalassemia ward in Cipto Mangunkusumo General Hospital in Jakarta after fully informed consent which conforms to the standards applied in Indonesia
Clinical Trial Registration
This research does not involve any clinical trials
Author Contributions
SKUF: experimental laboratory work, data collection and analysis, visualization, writing-original draft, approval of the final version; SKUF conducted this study as part of her dissertation at the University of Sydney.
HJF: conceptualization, methodology, blood sampling in Indonesia and transport to Australia, writing-review, approval of the final version, supervision, project administration; HJF designed the study concept for the collaboration between the Study Program Biomedical Sciences, Faculty of Medicine, Universitas Indonesia, and the Chemistry Department, University of Sydney.
References
- Stevens ML. Fundamentals of Clinical Hematology. Philadelphia: WB Saunders Co Ltd.; 1997
- Weatherall D, Porter JB, Clegg JB, Rees DC. Why are hemoglobin F levels increased in HbE/beta-thalassemia? Blood 1999;94:3199-3204
CrossRef - Brittain T. Molecular aspects of embryonic hemoglobin function. Mol Asp Med. 2002;23:293-342
CrossRef - Rachmilewitz EA, Rund D. New trends in the treatment of beta-thalassemia. Crit Rev Oncol Hematol. 2000;33:105-118
CrossRef - Thom CS, Dickson CF, Gell DA, Weis MJ. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harb Perspect Med. 2013;3:a011858
CrossRef - Origa R. β-Thalassemia. Genet Med 2017;19(6):609-619
CrossRef - Lie-Injo LE, Cai SP, Wahidiyat I, Moeslichan S, Lim KL, Evangelista L, Doherty M, Khan YW. Beta-thalassemia mutations in Indonesia and their linkage to different haplotypes. Am J Hum Genet. 1989;45:971-975
- Sofro AS. Molecular pathology of beta-thalassemia in Indonesia. Southeast Asian J Trop Med Pub Health 1995;26:221-224
- Setianingsih I, Williamson R, Marzuki S, Harahap A, Tamam M, Forrest S. Molecular basis of beta-thalassemia in Indonesia: application to prenatal diagnosis. Mol Diagn. 1998;3:11-19
CrossRef - Setianingsih I, Williamson R, Daud D, Harahap A, Marzuki S, Forrest S. Phenotypic variability of Filipino beta(o)-thalassemia/HbE patients in Indonesia: application to prenatal diagnosis. Am J Hematol. 1999;62:7-12
CrossRef - Dasgupta J, Sen U, Choudhury D, Datta P, Chakrabarti A, Chakrabarti SB, Dattagupta JK. Crystallization and preliminary X-ray structural studies of hemoglobin A2and hemoglobin E, isolated from the blood samples of β-thalassemic patients. Biochem Biophys Res Commun. 2003;303:619-623
CrossRef - Perutz MF. Stereochemistry of Cooperative Effects in Haemoglobin, Haem-Haem Interaction and the Problem of Allostery. Nature 1970;228:726-734
CrossRef - Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics 1999;15:327-332
CrossRef - Aitken JB, Austin CJD, Hunt NH, Ball HJ, Lay PA. The Fe-heme structure of met-indoleamine 2,3-dioxygenase-2 determined by X-ray absorption fine structure. Biochem Biophys Res Comm. 2014;450:25–29.
CrossRef - Rich AM, Armstrong RS, Ellis PJ, Freeman HC, Lay PA. Determination of iron-ligand bond lengths in horse heart met- and deoxymyoglobin using multiple-scattering XAFS analyses. Inorg Chem. 1998;37:5743-5753
CrossRef - Rich AM, Armstrong RS, Ellis PJ, Lay PA. Determination of Fe-ligand bond lengths and Fe-N-O bond angles in horse heart ferric and ferrous nitrosylmyoglobin using multiple-scattering XAFS analyses. J Am Chem Soc. 1998;120:10827-10836
CrossRef - Witting PK, Mauk AG, Lay PA. Role of Tyrosine-103 in Myoglobin Peroxidase Activity: Kinetic and Steady-State Studies on the Reaction of Wild-Type and Variant Recombinant Human Myoglobins with H2O2. Biochemistry 2002;42:11495-11503
CrossRef - Arnone A, Perutz MF. Structure of inositol hexaphosphate human deoxyhaemoglobin complex. Nature 1974;249:34-36
CrossRef - Nohl H, Stolze K. Formation of Methemoglobin and Free Radicals in Erythrocytes. In: A Sigel, H Sigel (Eds.) Metal Ions in Biological Systems, Vol. 36. Interrelations between free radicals and metal ions in life processes. NY, Basel, HongKong: M. Dekker, Inc.; 1999: pp. 289-307
CrossRef - Dickson CF, Rich AM, D’Avigdor WMH, Collins DAT, Lowry JA, Mollan TL, Khandros E, Olson JS, Weiss MJ, Mackay JP, Lay PA, Gell DA. ɑ-Hemoglobin-stabilizing Protein (AHSP) Perturbs the Proximal Heme Pocket of Oxy-hemoglobin and Weakens the Iron-Oxygen Bond. J Biol Chem. 2013;288(27):19986–20001
CrossRef - Collman JP, Gagne RR, Reed CA, Robinson WT, Rodley GA. Structure of an iron (II) dioxygen carrying hemeproteins. Proc Nat Acad Sci. USA 1974;71:1326-1329
CrossRef - Perutz MF. Regulation of oxygen affinity of hemoglobin: influence of structure of the globin on the heme iron. Ann Rev Biochem. 1979;48:327-386
CrossRef - Udyaningsih-Freisleben SK, Kurniati V, Prasetyo PB, Handayani S, Adhiyanto C, Soegianto RR, Pudiantari R, Munthe BG, Ramelan W, Freisleben HJ. Isolated erythrocyte membranes of transfusion-dependent and non-transfused thalassemia patients in Jakarta – investigated by electron paramagnetic resonance spectroscopy, BioFactors 2003;19:87-100
CrossRef - Freisleben SKU, Hidayat J, Freisleben HJ, Poertadji S, Kurniawan B, Na Peng Bo, Handayani S, Wahidiyat PA, Soegianto RR. Plasma lipid pattern and red cell membrane structure in -thalassemia patients in Jakarta, Med J Indones. 2011;20(3):178-184
CrossRef - Laksmitawati DR, Handayani S, Udyaningsih-Freisleben SK, Kurniati V, Adhiyanto C, Hidayat J, Kusnandar S, Dillon HSD, Munthe BG, Wirawan R, Soegianto RR, Ramelan W, Freisleben HJ. Iron status and oxidative stress in ß-thalassemia patients in Jakarta, BioFactors 2003;19:53-62
CrossRef - Caughey WS, Watkins JA. Oxy radical and peroxideformation by hemoglobin and myoglobin. In: RA Greenwald (Ed.) Handbook of Methods for Oxygen Radical Research. Boca Raton: CRC Press; 1986: pp. 95-103
- Udyaningsih-Freisleben SK. XAS and RR Structural Analysis of Hemoglobin and EPR Spectroscopic Labelling of Red Blood Cell Membranes Isolated From Thalassemia Patients in Jakarta, Indonesia. PhD Thesis, University of Sydney, Australia; 2003
- Ellis PJ, Freeman HC. XFIT – an Interactive EXAFS Analysis Program. J Synchrotron Rad 1995;2:190-195
CrossRef - Rich AM. Determination of Fe-ligand bond lengths and angles in heme proteins using X-ray absorption spectroscopy. PhD Thesis, University of Sydney, Australia; 1997
- Springer BA, Sligar SG, Olson JS, Phillips GN, Jr. Mechanism of ligand recognition in myoglobin. Chem Rev. 1994;94:699-714
CrossRef - Antonini E, Brunori M. Hemoglobin and myoglobin in their reactions with ligands. In: A Neuberger, EL Tatum (Eds.) Frontiers of Biology, Vol. 21. Amsterdam, London; North-Holland Publishing Company: 1971
- Pin S, Alpert B, Congiu-Castellano A, Della Longa S, Bianconi A. X-ray absorption spectroscopy of hemoglobin. Methods Enzymol. 1994;232:266-292
CrossRef - Brucker EA. Genetically crosslinked hemoglobin: a structural study. Acta Cryst. 2000;D56:812-816
CrossRef - Shiver DF, Atkins PW, Langford CH. Inorganic Chemistry Edn. 2, Oxford, UK; Oxford University Press: 1994
- Park S-Y, Yokoyama T, Shibayama N, Shiro Y, Tame JRH. 1.25 Å Resolution Crystal Structures of Human Haemoglobin in the Oxy, Deoxy and Carbonmonoxy Forms. J Mol Biol. 2006;360:690–701
CrossRef - Bianconi A, Congiu-Castellano A, Dell’Ariccia M, Giovannelli A, Morante S, Burattini E, Durham PJ. Local Fe site structure in the tense-to-relaxed transition in carp deoxyhemoglobin: a XANES (x-ray absorption near edge structure) study. Proc Natl Acad Sci. USA 1986;83:7736-7740.
CrossRef - Paoli M, Liddington R, Tame J, Wilkinson A, Dodson G. Crystal structure of T state haemoglobin with oxygen bound to all four haems. J Mol Biol. 1996;256:775-792
CrossRef - Reddy LR, Reddy KS, Surrey S, Adachi K. Role of Hydrophobic Amino Acids at ß85 and ß88 in Stabilizing F Helix Confirmation of Hemoglobin S. J Biol Chem 1996;271:24564-24568
CrossRef - Williamson D. The unstable hemoglobins. Blood Rev 1993;7:146-163
CrossRef - McArthur KM, Davies M. Detection and reactions of the globin radical in haemoglobin. Biochem Biophys Acta 1993;1202:173-181
CrossRef - Rees DC, Clegg JB, Weatherall DJ. Is Hemoglobin Instability Important in the Interaction Between Hemoglobin E and ß Thalassemia? Blood 1998; 92(6): 2141-2146
CrossRef - Hirsch RE, Sibmooh N, Fucharoen S, Friedman JM. HbE/ß-Thalassemia and Oxidative Stress: The Key to Pathophysiological Mechanisms and Novel Therapeutics. Antioxid Redox Signal 2017; 26(14): 794-813
CrossRef - Ondei LS, Estevão IF, Rocha MI, Percário S, Souza DR, Pinhel MA, Bonini-Domingos CR. Oxidative stress and antioxidant status in beta-thalassemia heterozygotes. Rev Bras Hematol Hemoter. 2013;35(6):409-413
CrossRef - Jomouia W, Satthakarn S, Panyasai S. Molecular understanding of unusual HbE-β+-thalassemia with Hb phenotype similar to HbE heterozygote: simple and rapid differentiation using HbE levels. Ann Med 2023;55(2):2267054
CrossRef - Rees DC. Hemoglobin F and hemoglobin E/beta-thalassemia. J Pediatr Hematol Oncol 2000; 22: 567–572
CrossRef - Strader MB, Kassa T, Meng F, Wood FB, Hirsch RE, Friedman JM, Alayash AI. Oxidative instability of hemoglobin E (β26 Glu-Lys) is increased in the presence of free α subunits and reversed by α-hemoglobin stabilizing protein (AHSP): Relevance to HbE/β-thalassemia. Redox Biology 2016;8:363–374
CrossRef - Samuel PP, White MA, Ou WC, Case DA, Phillips GN Jr, Olson JS. The Interplay between Molten Globules and Heme Disassociation Defines Human Hemoglobin Disassembly. Biophys J 2020;118:1381–1400
CrossRef - Wang D, Zhang H, Yang Z, Su W, Dou Y, Xu Y. Case report: A rare heterozygous Hb CS with heterozygous HbE in a family with thalassemia in China. Heliyon 2024; 10: e37858
CrossRef

