
Manuscript accepted on :15-04-2024
Published online on: 03-06-2024
Plagiarism Check: Yes
Reviewed by: Dr. Neeta Rai and Dr. Pranjal Gujarathi
Second Review by: Dr. Abhishek Raj
Final Approval by: Dr Ayush Dogra
Alok Kumar Moharana , Tapas Kumar Mohapatra, Rudra Narayan Dash , and Bharat Bhusan Subudhi*
, Tapas Kumar Mohapatra, Rudra Narayan Dash , and Bharat Bhusan Subudhi*
Drug Development and Analysis Lab, School of Pharmaceutical Sciences, Siksha ‘O’ Anusandhan Deemed to be University, Bhubaneswar, India.
Corresponding Author E-mail: bbsubudhi@soa.ac.in
DOI : https://dx.doi.org/10.13005/bpj/2906
Abstract
Chikungunya virus (CHIKV) infection was previously found to be inhibited by MBZM-N-IBT both in vitro and in vivo. To further assess its suitability for in vivo application, toxicity and pharmacokinetics were investigated. It showed no acute toxicity orally with an estimated LD50 of more than 5000 mg/kg in rats. While it showed toxicity at 1000 mg/kg in the chronic toxicity study, it was better tolerated at 500 mg/kg by rats. At 50 mg/kg, it was found to be safe in a 9-month study. A pharmacokinetic study revealed Tmax less than the gastric emptying time. High plasma protein binding supported its higher elimination half-life. In silico analysis predicted 22 metabolites. The majority of these metabolites fall in OECD class 5 and support the low toxicity of MBZM-N-IBT.
Keywords
Antiviral; MBZM-N-IBT; Pharmacokinetic; Toxicity
Download this article as:| Copy the following to cite this article: Moharana A. K, Mohapatra T. K, Dash R. N, Subudhi B. B. Pharmacokinetic and Safety Evaluation of MBZM-N-IBT, A Lead Against Chikungunya Virus. Biomed Pharmacol J 2024;17(2). |
| Copy the following to cite this URL: Moharana A. K, Mohapatra T. K, Dash R. N, Subudhi B. B. Pharmacokinetic and Safety Evaluation of MBZM-N-IBT, A Lead Against Chikungunya Virus. Biomed Pharmacol J 2024;17(2). Available from: https://bit.ly/3ySuIFg |
Introduction
Infection by Chikungunya virus (CHIKV) is categorised by WHO as a “neglected tropical disease” (WHO, 2014)1. Although it has a tropical origin, it is no longer limited to tropical areas. Gradually, it has become a global pathogen in many countries with different climatic conditions2,3. Although no specific antiviral is approved for its management, there have been continued efforts to identify potential drug candidates for further development as antiviral for CHIKV4-9. Our group recently patented MBZM-N-IBT (Figure 1) for its in vitro effects against CHIKV infection (Indian Patent No. 347450). Chemically it is 1-[(2-methyl benzimidazole-1-yl) methyl]-2-oxo-indolin-3-ylidene] amino] thiourea10. Unlike the prior art in its chemical class (methisazone), it was reported as a potent inhibitor of CHIKV10.
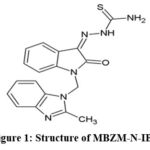 |
Figure 1: Structure of MBZM-N-IBT |
In addition to anti-CHIKV properties, it was shown as a potent inhibitor of the Herpes Simplex Virus (HSV1) in vitro11. The effects against both RNA (CHIKV) and DNA (HSV) virus suggest its broad spectrum of action and potential for further progress as an antiviral candidate. Pharmacokinetic study plays a vital role in drug development as it helps in the prediction of efficacy and toxicity-related events. Thus, to better understand the toxicity and the effectiveness of MBZM-N-IBT in vivo, it is necessary to assess its pharmacokinetics through a rapid, simple, and effective bio-analytical method12. MBZM-N-IBT is a chemical substance and may cause acute toxicity within 24 hours following single-dose administration. When continuously administered, it could lead to chronic toxicity in the living system when used over a more extended period of time.13,14. A key criterion to assess acute toxicity is the determination of the lethal dose (LD50), which is defined as the dose that kills 50% of the population of test animals.
Furthermore, it appears as a guiding dose for in vivo efficacy and toxicity studies15. Accordingly, the LD50 was estimated in the present study before assessing the chronic toxicity. In silico models have been widely used during the toxicology analysis16-18 to reduce safety-related attrition in drug development. So, the metabolites of MBZM-N-IBT were predicted through in silico models, and their potential toxicity was assessed to support the findings of in vivo toxicology. With these thoughts in mind, the goal of the current investigation was to evaluate both the toxicity and pharmacokinetics of MBZM-N-IBT to assess its potential for further progress as an antiviral candidate.
Material and Methods
Chemicals and Reagents
MBZM-N-IBT was synthesized and purified by column chromatography following our established protocol10. The purity (˃98%) was monitored by HPLC. Metformin, Metronidazole, Ranitidine, Paracetamol, Phenobarbital, Cefixime, Salicylic acid, Cetrizine, omeprazole, and Losartan were all obtained from industry with high purity (˃95%). The HPLC-grade solvents, such as acetonitrile, water, and methanol, have been purchased from Merck, India. Drugs/test compounds were added to 1% carboxy methyl cellulose (CMC) in distilled water at room temperature, and fresh crude suspensions were prepared.
Animals and Ethical Statement
The Institutional Animal Ethics Committee (IAEC) of the School of Pharmaceutical Sciences of Siksha O Anusandhan (Deemed to be University), Bhubaneswar, India, (Reg. no. 1171/PO/Re/S/08/ CPCSEA) granted its approval to the experimental protocol. The Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) approved all experiments and supervised the way they were conducted. To evaluate acute and chronic toxicity, healthy Wistar albino rats of either sex, around 150-200g, were used. They were kept in polyacrylic cages with 12-hour light/dark cycles at optimal temperatures of 25±2°C and 60±5% relative humidity. The animals had complete access to commercially available food and water.
Animal grouping and husbandry
Animals were grouped into subgroups according to the experiments after seven days of acclimation. For the acute toxicity study, animals were divided into 4 groups arbitrarily (n=3, female rats). For the chronic toxicity study, animals (both male and female rats) were randomly divided according to the dose and the time limit of the experiment. The in vivo pharmacokinetic study was performed by taking albino rats at each time point (n=6). All of the animals were housed in polypropylene cages with stainless steel covers that contained water bottles and feeders.
Dosing
MBZM-N-IBT was mixed with 1% carboxy methyl cellulose to achieve the desired concentration. Dosing formulations were stored at 4ºC. The formulations were brought to room temperature and thoroughly mixed before administration.
Acute oral toxicity of MBZM-N-IBT
Following the OECD-423 guidelines for testing chemicals (OECD, 2000)19,20, this experiment was carried out to estimate the LD50 of MBZM-N-IBT. Following an overnight fast of approximately 15 to 16 hours, all the animals were given a single oral dose of MBZM-N-IBT with unrestricted access to water. The dose volume was 10 mL/kg. The starting dose was 50 mg/kg body weight. The animals were next examined with 300 mg/kg and 2000 mg/kg single oral doses. The toxic impact of each and every experimental animal was observed for the entire day. For the first 4h of daily administration, animals were critically monitored for any signs of toxicity. The changes in behavior, lethargy, appearance, salivation, illness, body weight, and mortality were examined during this period. The animals were further observed for 14 days.
Chronic oral toxicity of MBZM-N-IBT
Chronic toxicity was conducted as per the OECD-452 guidelines (OECD, 2008)21. As per the recommendations of this guideline, the highest dose that can be administered is 1000 mg/kg. The animals were divided into 4 groups arbitrarily. Each group contained male and female rats (n=20 for each sex). The Group-I (normal control) rats received 1% CMC. Group II, III, and IV rats received oral MBZM-N-IBT at 1000 mg/kg, 500 mg/kg, and 50 mg/kg doses. All doses were administered once per day with a fixed schedule. Animals were observed for gross behavioral, neurological, and autonomic effects. Also, changes in eyes, skin/fur, salivation, sleep, diarrhea, coma, and mortality were noted twice daily throughout the testing period. On a weekly basis, the rats’ body weight was monitored. After the completion of the period, ketamine was injected intraperitoneally, and rats were euthanized. For histological analysis, vital organs like the liver, heart, lung, and kidney were carefully excised, weighed, and preserved in 10% buffered formalin.
Bioanalytical method development and Pharmacokinetics of MBZM-N-IBT
A binary gradient HPLC (Shimadzu, Japan) was employed, along with an LC-20AD pump and SPD-M20A prominence diode array detector. Samples were injected into a KROMASIL PROCHROME C18 analytical column (4.6 × 150mm, 5μm) using a Hamilton SYR 25μL syringe after being filtered through a syringe-driven nylon membrane filter (0.22µ, 13mm). With an isocratic flow rate of 1.0mL/min and water methanol (50: 50 v/v) as the mobile phase, the estimation of MBZM-N-IBT was carried out. The mobile phase was degassed in an ultrasonic bath after being filtered through a Millipore vacuum filter system with a 0.45μm membrane filter. Each injection ran for ten minutes. At 360nm, the analytes were observed. At ambient temperature, all experiments were carried out. Following ICH guidelines Q2 (R1), a reversed-phase HPLC analytical technique was developed and validated for the quantitative determination of MBZM-N-IBT from bulk22. The linearity, accuracy, precision, LOD, and LOQ were determined. Again, this method was verified and re-validated for assay from rat plasma matrix following USFDA guidelines for bio-analytical method validation23,24.The calibration curve was used to estimate MBZM-N-IBT in plasma. The in vivo Pharmacokinetic study was performed by taking three albino rats at each time point (n=6). Animals were orally administered with MBZM-N-IBT 50 mg/kg body weight. At appropriate time points (0.5, 1, 1.5, 2, 4, 8, 12, 18, and 24 hr), the animals were euthanized under light ether anesthesia, and blood was obtained through the cardiac puncture, which was then collected into a pre-treated vial containing EDTA. Following blood collection, the samples were centrifuged at 15,000 rpm for 15 minutes at 4°C to separate the plasma. Then, clear supernatants were collected and subjected to deproteinization (Acetonitrile).
The collected samples were preserved at -80°C until they were analyzed. Different pharmacokinetic parameters have been determined, including AUC, AUMC, MRT, Cmax, Tmax, and T1/2.
Protein binding study
Plasma protein binding (PPB) was evaluated using a modified version of a previously reported procedure25. Drugs with reported PPB values ranging from 0% to 99% were selected for the research. Metformin, metronidazole, ranitidine, paracetamol, phenobarbital, cefixime, salicylic acid, cetirizine, omeprazole, and losartan were all obtained from industry with high purity (˃95%). An isocratic mobile phase, composed of a 0.9% sodium chloride solution at pH 7.0, was employed, and it was run at a flow rate of 0.5 mL/min. The study was performed using a chiral pack HSA column (150×4 mm, 5µm). A significant correlation was achieved by generating a graph that compared the derived capacity factors with the reported (known) PPB percentages.
In silico analysis of metabolites and their LD50
The OECD QSAR Toolbox, owned by the OECD and the European Chemical Agency, is a tool used for the assessment of chemicals and the mechanistic basis of their toxicology26. The classical interface of QSAR tool box4.6, 2023 was used for the prediction of metabolite and their toxicity. MBZM-N-IBT structure was used as input. Several metabolism simulators were used to predict the metabolites, including in vivo Rat metabolism, autoxidation, and hydrolysis (acidic, basic, and neutral). These metabolites were utilized as inputs for predicting acute toxicity using the OECD QSAR toolbox, which aids in categorizing chemicals into specific groups.
Statistics
All data are expressed as mean±SEM (Standard Error Mean) and were analyzed by the XLSTAT software using one-way ANOVA followed by Bonferroni multiple comparison tests (p˂0.001).
Results
MBZM-N-IBT showed no acute oral toxicity
After administration of different single oral doses (50 mg/kg, 300 mg/kg, and 2000 mg/kg) of MBZM-N-IBT, the animals were observed for the first 4h and daily up to 14 days. There were no behavioral changes or mortality (Table 1) with different doses of MBZM-N-IBT. As a result, it was classified as “Category 5” or “unclassified” as per the Globally Harmonized System (GHS) for chemical classification. Accordingly, 5000 mg/kg was considered as the LD50.
Table 1: Acute oral toxicity observation table
|
Step |
Dose (mg/kg) |
Number of Animals |
Number of moribund/ deceased Animals |
Subsequent dead animal |
|
1 |
50 |
3 |
0 |
0 |
|
2 |
50 |
3 |
0 |
0 |
|
3 |
300 |
3 |
0 |
0 |
|
4 |
300 |
3 |
0 |
0 |
|
5 |
2000 |
3 |
0 |
0 |
|
6 |
2000 |
3 |
0 |
0 |
No significant chronic toxicity was found on chronic use of MBZM-N-IBT (50 mg/kg)
MBZM-N-IBT (50 mg/kg) did not affect the food and water intake of animals
Physiological changes (Table 2) were observed following the administration of MBZM-N-IBT at a dose of 1000 mg/kg/day (the highest permissible dose) and 500 mg/kg/day. Rats treated daily with 500 mg/kg showed no abnormality for up to 28 days (Table 2). Beyond 42 days, their food/water intake capacity was observed to decrease (Figure 2). A simultaneous decrease in body weight was also observed, and it became significant after 60 days. Rats treated with 50 mg/kg/day showed no abnormality even after 120 days of administration (Table 2). The average food intake and water consumption (Figure 2) were observed to be average at this dose. Although after 270 days, most of the parameters were normal (Table 2), It exhibited mild sedation, along with an 11% reduction in body weight. The colour and consistency of the stool were yellowish and loose. The dose at 1000mg/kg/day showed toxicity (Figure 3) in rats after 28 days of continuous administration. Since mortality was found with the highest dose, this was discontinued.
Table 2: Behavioural changes in the animal during chronic toxicity studies following treatment with MBZM-N-IBT
|
Parameters |
1000 mg/kg |
500 mg/kg |
50 mg/kg |
50 mg/kg |
|
28 days |
60 days |
120days |
270 days |
|
|
Condition of fur |
Abnormal |
Normal |
Normal |
Normal |
|
Skin |
Normal |
Normal |
Normal |
Normal |
|
Subcutaneous swelling |
Nil |
Nil |
Nil |
Nil |
|
Eyes dullness |
Present |
Nil |
Nil |
Present |
|
Breathing abnormalities |
Present |
Nil |
Nil |
Nil |
|
Food intake |
Decreased after 7days |
Normal till 28 days |
Normal |
Normal |
|
Colour/consistency of feces |
Abnormal |
Normal |
Normal |
Yellowish/loose |
|
Alertness |
Abnormal after one week |
Normal |
Normal |
Normal |
|
Convulsions |
Not observed |
Not observed |
Not observed |
Not observed |
|
CNS |
Sedation |
Sedation |
Sedation |
Sedation |
|
Body temperature |
Normal |
Normal |
Normal |
Normal |
|
Bodyweight |
Decreased after one week |
Decreased after 42 days |
Normal |
Reduced by 11% |
|
Grooming |
Absent |
Present |
Present |
Present |
|
Death |
3 animals were found dead during the testing period |
No death after 60 days |
No death |
No death |
 |
Figure 2: Mean food and water intake of animals. Food (a and b) and water (g and h) intake of animals treated with 1000 mg/kg up to 28 days. |
Histopathology revealed safety of MBZM-N-IBT (50 mg/kg)
The rat liver tissues (Figures 3a, 4a, and 5a) from the control group showed normal hepatic morphology, but MBZM-N-IBT (1000 mg/kg for 28 days) treated rat showed activated kupffer cells, infiltration of lymphocytes at the portal/central vein, sinusoidal obstruction, fibrous connective tissue and necrotic plaques (Figure 3b). In the case of MBZM-N-IBT (500mg/kg for 60 days), hepatocytes were loosely arranged, and vacuolar degeneration of hepatocytes was observed (Figure 4b). Following MBZM-N-IBT (50 mg/kg for 270 days) treatment, the liver showed alteration of cytoarchitecture (the arrowhead points to hepatic lobules, where the trabecular structure appears slightly blurred, while the remaining lobules exhibit a distinct blurring). Empty vacuolar spaces were seen in the cytoplasm of some cells. Kupffer cells were additionally detected along the sinusoidal walls (Figure 5b). The kidney tissues (Figures 3c, 4c, and 5c) from the normal control group showed the presence of normal renal corpuscle and kidney tubules. The MBZM-N-IBT (1000 mg/kg for 28 days) treated rat showed the presence of granular cast, focal proximal tubular epithelial necrosis, and unusual morphological pattern with the degeneration of the renal corpuscles (Figure 3d). Kidney tissues from MBZM-N-IBT (500 mg/kg for 60 days) treated rats showed cellular cast and Protein and reduction in the renal corpuscles (Figure 4d). The section of the kidney showed a normal appearance (the arrowhead highlights that the collecting tubules are lined with a simple cuboidal epithelium, and there is adjacent interstitial tissue) in case of treatment at 50 mg/kg/day for 270 days (Figure 5d). The stomach tissues (Figures 3e, 4e, and 5e) from the normal control group showed the presence of apparent normal mucosa and submucosa and thickened mucosal layer. Stomach tissues from MBZM-N-IBT (1000 mg/kg for 28 days) treated rats showed minor epithelial ulceration covered by fibrin entangling neutrophils involving the reduction of gastric mucosal thickening (Figure 3f). Whereas 500 mg/kg for 60 days (Figure 4f) and 50 mg/kg for 270 days (Figure 5f) treatment showed no alteration in stomach cytoarchitecture of the rats. The lung tissues (Figures 3g, 4g, and 5g) from the normal control group showed polygonal alveoli and blood vessels. The presence of thin interalveolar septa and well-inflated alveoli indicate normal lung architecture. The MBZM-N-IBT (1000 mg/kg for 28 days) treated rat showed collapsed alveoli, suggesting severe alveolar damage (Figure 3h).
In contrast, rats treated with a lesser dose (500mg/kg for 60 days) showed slight interalveolar septa thickening. Whereas this suggests a relatively low level of toxicity, isolated focal regions with inflammatory cell infiltration and the appearance of clogged, thickened blood vessels indicate adverse effects on the lungs with continued treatment (Figure 4h). Following MBZM-N-IBT (50mg/kg for 270 days) treatment, normal intrapulmonary bronchioles could be seen (Figure 5h). The cardiac tissues (Figures 3i, 4i, and 5i) from the normal control group showed no necrosis throughout the musculature, no foci of cellular infiltration, normal nuclei, and normal cytoplasm. However, treated rats with MBZM-N-IBT (1000 mg/kg for 28 days) displayed distinct foci of cellular infiltration, having large nuclei and specific areas that showed general cellular ultrastructure disruption (Figure 3j). The rat treated with MBZM-N-IBT (500mg/kg for 60 days) displayed loss of nuclei, indicating dead cells and internalization of large nuclei indicative of the myocyte regeneration process (Figure 4j). The histological section of the myocardium (Figure 5j) showed a normal appearance (arrowhead indicates muscle fiber and blood vessel) in the case of MBZM-N-IBT (50mg/kg for 270 days). The brain tissues (Figures 3k, 4k, and 5k) from the normal control group showed the cerebral cortex with regular ependyma of the ventricle, pyramidal cells, and perivascular space. The MBZM-N-IBT (1000mg/kg for 28 days) treated rat showed blood vessels dilated with increased perivascular spaces and intervening edema in the form of vacuoles in the brain substance and ischaemic necrosis (Figure 3l). Brain tissues from MBZM-N-IBT (500 mg/kg for 60 days) treated rats showed pyknotic nuclei and increased perivascular space with hemorrhage and vacuoles in the brain (Figure 4l). Meanwhile, the brain tissue (Figure 5l) showed a normal appearance (arrowhead indicates different sizes and shapes of nerve cells and nerve fibers) in the case of MBZM-N-IBT (50 mg/kg for 270 days). This suggests that the dose of 50 mg/kg/day is relatively safe for oral administration.
 |
Figure 3: Histopathology of rat tissues after the administration of MBZM-N-IBT (1000 mg/kg) for 28 days. |
 |
Figure 4: Histopathology of rat tissues after the administration of MBZM-N-IBT (500 mg/kg) for 60 days. |
 |
Figure 5: Histopathology of rat tissues after the administration of MBZM-N-IBT (50 mg/kg) for 270 days. |
Bioanalytical Method Development and Pharmacokinetics of MBZM-N-IBT
A novel RP-HPLC (Reverse Phase High-Performance Liquid Chromatography) method was established and validated in compliance with the ICH (International Council for Harmonization) guidelines, specifically ICH Q2 (R1). All the parameters were validated, and the results are shown in Table 3. After this, the method was verified according to USFDA guidelines for bio-analytical method validation. Specificity was confirmed by the absence of peaks at the retention time of MBZM-N-IBT from the plasma. The calibration curve was generated by adding a known quantity of MBZM-N-IBT to 500 µL of rat plasma, resulting in concentrations of 2, 4, 6, 8, and 10 µg/mL. MBZM-N-IBT in rat plasma was quantified by measuring the analyte’s response against the calibration curve. The concentration of MBZM-N-IBT in six replicates of plasma spiked with 4, 6, and 8 µg/mL of MBZM-N-IBT was evaluated using HPLC in a single day and repeated on three separate days to evaluate accuracy and precision. In accordance with FDA guidelines, the acceptance criteria for accuracy were set at 85-115%, and the coefficient of variation values (CV) was required to be ˂ 15%, except at the Lower Limit of Quantification (LLOQ), where accuracy was acceptable within the range of 80-120%, and the CV ≤ 20% (Table 4). The pharmacokinetic plasma profiles of MBZM-N-IBT after 50 mg/kg oral administration are depicted in Figure 6, and pharmacokinetic parameters are summarized in Table 5. The estimated maximum plasma concentration (Cmax) was 1.079±0.069 μg/mL, and the duration to achieve the maximum plasma concentration (Tmax) was 2h. This shows that absorption of MBZM-N-IBT is not slow and is within the normal gastric emptying time of 2-4 hours. The apparent elimination half-life (t1/2) was calculated as 8.120±0.069h, which indicates that more than 40h (five half-lives) may be required for complete clearance of MBZM-N-IBT from systemic circulation.
Table 3: Validation parameter for analysis of MBZM-N-IBT from bulk
|
Linearity |
r2=0.999 |
0.5-100 µg/mL |
||
|
Accuracy (%±SD) |
100.16 ± 0.557 |
|||
|
Precision (RSD %) |
Intra-day |
Inter-day |
||
|
Day 1 |
Day 2 |
Day 3 |
||
|
0.722 |
0.55 |
0.57 |
1.31 |
|
|
LOQ |
0.150 µg/mL |
|||
|
LOD |
0.050 µg/mL |
|||
Table 4: Validation of analytical method for analysis of MBZM-N-IBT from plasma.
|
Linearity |
r2=0.999 |
2-10 µg/ml |
|||
|
Accuracy (%±SD) |
LQC |
96.7±0.094 |
|||
|
MQC |
97.5±0.113 |
||||
|
HQC |
93.9±0.244 |
||||
|
Precision |
Intra-day |
Inter-day |
|||
|
Day 1 |
Day 2 |
Day 3 |
|||
|
LQC |
2.55 |
3.36 |
2.85 |
3.31 |
|
|
MQC |
1.93 |
3.11 |
3.80 |
2.27 |
|
|
HQC |
3.24 |
1.92 |
3.29 |
2.89 |
|
|
LLOQ |
2 µg/ml |
||||
|
LOD |
0.4 µg/ml |
||||
Table 5: Pharmacokinetic parameters of MBZM-N-IBT
|
Parameter |
Result |
|
AUC (µg/mL h) |
25.604±0.123 |
|
AUMC(µg/mL h2) |
298.346±0.412 |
|
MRT (h) |
11.652±0.040 |
|
Cmax (µg/mL) |
1.709±0.069 |
|
Tmax (h) |
2 |
|
Kel (h2) |
0.085±0.031 |
|
t1/2 |
8.120±0.069 |
*mean±SD (n=6)
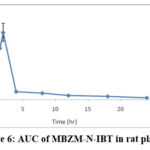 |
Figure 6: AUC of MBZM-N-IBT in rat plasma. |
Plasma protein binding of MBZM-N-IBT
The HPLC method estimated Plasma protein binding using a human-HSA chiral column. The capacity factor measures the retention of drugs in the column. Accordingly, the capacity factor of drugs while using a human-HSA chiral column represents its affinity towards human serum albumin. Since this is the major plasma protein, it’s binding correlates well with drugs’ in vivo plasma protein binding. To corroborate this, various drugs with reported plasma protein binding in the range of 1-99% were considered. Their capacity factors were determined (Table 6). A significant correlation (r2=0.994) was found between the capacity factor and the known PPB% of these drugs. Accordingly, this showed a linear regression (Y= 0.008X+0.144, Figure 7). The modified capacity factor (K´/(K´+1)) of MBZM-N-IBT was determined to be 0.94. Accordingly, its PPB% was found to be 99.5 (Table 6). This suggests that MBZM-N-IBT has a high plasma protein binding capacity.
Table 6: Plasma protein binding % of different drugs
|
Drug |
Reported PPB % |
Rt |
K´ |
K´/(K´+1) |
Measured PPB % |
|
Metformin27 |
0 |
3.161 |
0.161 |
0.138 |
0.00 |
|
Metronidazole28 |
10 |
3.546 |
0.244 |
0.196 |
12.5 |
|
Ranitidine29 |
15 |
4.032 |
0.482 |
0.325 |
22.625 |
|
Paracetamol30 |
25 |
4.494 |
0.571 |
0.363 |
27.375 |
|
Phenobarbital31 |
45 |
6.44 |
1.027 |
0.506 |
45.25 |
|
Cefixime32 |
67 |
2.798 |
2.436 |
0.708 |
70.05 |
|
Salicylic acid33 |
85 |
15.610 |
8.974 |
0.899 |
94.375 |
|
Cetrizine34 |
93 |
28.662 |
16.834 |
0.943 |
99.875 |
|
Omeprazole35 |
95 |
40.638 |
24.414 |
0.96 |
102 |
|
Losartan36 |
99 |
52.971 |
32.985 |
0.970 |
103.25 |
|
MBZM-N-IBT |
– |
51.962 |
17.248 |
0.94 |
99.5 |
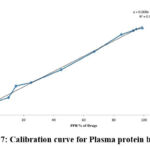 |
Figure 7: Calibration curve for Plasma protein binding |
Metabolites of MBZM-N-IBT showed no toxicity.
No metabolite was found for metabolism in the autoxidation and dissociation simulator, indicating the molecule’s stability. A simulator for hydrolysis under neutral conditions predicted 5 metabolites. Under acidic and basic conditions, the hydrolysis simulator predicted 12 and 13 metabolites, respectively. In the in vivo rat simulator, 13 metabolites were predicted. After combining the results of all these simulators, 22 unique metabolites were predicted for MBZM-N-IBT (Table 7). While 21 metabolites were expected to be OECD class five chemicals with very high LD50, one metabolite was categorized as OECD class 4 with a potential LD50 of 681mg/kg. This indicates that the metabolites of MBZM-N-IBT are not likely to be toxic.
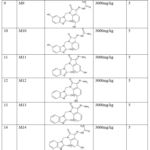 |
Table 7: Predicted Metabolites of MBZM-N-IBT. |
Discussion
MBZM-N-IBT is an important antiviral lead. It has a broad spectrum of antiviral action and the potential to interfere in multiple ways against CHIKV and HSV replication6,11. However, it has been demonstrated to be effective in vivo at a dosage of 15 mg/kg against CHIKV infection in mice; further preclinical validation is necessary to justify its suitability as an antiviral candidate37. Hence, assessing its pharmacokinetics and toxicology is essential to encourage further in vivo validation.
An oral acute toxicity study, as per OECD-423 guidelines, revealed that rats had no acute toxicity during the monitoring period (14 days) (Table 1). Accordingly, the oral LD50 of the MBZM-N-IBT was calculated to be greater than 5000 mg/kg as per the OECD-423 guidelines. This suggested the acute oral safety of MBZM-N-IBT. In the chronic toxicity studies, MBZM-N-IBT administration at a high dose for 28 days (1000 mg/kg/day) led to a remarkable reduction in food intake and water consumption. Although this was normal in the first three weeks in animals treated with a moderate dose (500 mg/kg/day), in the later stages, there was a reduction in food intake and water consumption (Figure 2). At a lower dose (50 mg/kg/day), rats showed no significant change in food intake and water consumption for 270 days. Also, there were no signs of toxicity and behavioural changes (Table 2). However, on prolonged use, some reversible sedation was noted. Besides, there were some changes in stool and body weight (Table 2). In congruence with these, significant alterations were observed in the pathological study of the excised liver, lungs, kidney, stomach, heart, and brain (Figures 3 & 4). At the end of the 270-day study, some necrosis of excised cardiomyocytes at the sub-endocardium region in cardiac tissue was observed. In excised brain tissue, vascular congestion, and cellular infiltration were also marked (Figure 5).
Nevertheless, the pathological study of the excised liver, kidney, stomach, and lung tissue showed a normal appearance (Figure 5). The sedation on prolonged use suggests the ability to reach the brain. Nevertheless, till 120 days and beyond, it showed no effect on motor coordination, locomotion, and anxiety at a dose of 50 mg/kg. Since the antiviral application usually requires short-term drug administration, this study hints at the safety of this dose for further investigation.
The analytical and bio-analytical method validation parameters are shown in Tables 3 and 4. Following an oral dose of 50 mg/kg to rats, the AUC of MBZM-N-IBT was determined (Figure 6). It revealed that 2h is taken to achieve the highest systemic concentration. This shows that it is absorbed within 2h of oral administration. Considering the fact that the normal gastric emptying time is between 2-4h, the absorption rate of MBZM-N-IBT may be adequate.
Further, its elimination half-life was found to be 8 h (Table 5). Drugs with a half-life of 12-48 h are generally considered suitable for once-daily oral dosing38. Although the half-life of MBZM-N-IBT is less than this, it can remain in circulation for 5 half-lives or 40h. Hence, its suitability for once-daily oral dosing cannot be ruled out without further validation. Moreover, it has been demonstrated to be effective against CHIKV infection in mice with a once-daily oral dosing of 15 mg/kg37.
The protein binding was estimated using the HPLC method by measuring its affinity for human serum albumin attached to the stationary phase. To develop this method, the modified capacity of different drugs that represent their affinity for human serum albumin was correlated to their known PPB%. A high correlation (r2 = 0.994) suggested the suitability of the regression equation that was used for the calculation of PPB% (Figure 7). As shown in Table 6, the calculated PPB% was very close to that of the reported values for most of the drugs. Hence, 99.5% PPB was estimated for MBZM-N-IBT. This high PPB is in agreement with the relatively high elimination half-life (8h) and may support the longer duration of action.
Further, the metabolism of MBZM-N-IBT was predicted through in silico analysis using the OECD QSAR Toolbox. The reliability of the OECD QSAR toolbox has been demonstrated for the prediction of the acute toxicity of organic chemicals39. It is supported by the Organisation for Economic Co-operation and Development (OECD) as well as the European Chemicals Agency (ECHA). Using these tools, 22 unique metabolites (Table 7) were predicted. While 21 of these metabolites were non-toxic (OECD-class 5), one was predicted to have an LD50 of 681mg/kg (Table 7). Thus, like MBZM-N-IBT, the metabolites are also not likely to be toxic. Since these metabolites were mainly predicted in the rat simulator model, this may also support the safety in rats demonstrated in the chronic toxicity studies. However, further in vivo metabolite analysis is necessary to validate this.
Conclusion
The no-observed-adverse-effect level (NOAEL) of MBZM-N-IBT can be taken into consideration to be 50 mg/kg under the specified circumstances based on the single-dose oral acute toxicity study and the lack of substantial toxicity or mortality in the sub-chronic toxicity study. While the in vivo pharmacokinetics suggests adequate rates of absorption, elimination half-life (8h), and high PPB suggest a potentially longer duration of action in vivo. The predicted metabolites are not expected to be toxic and support the in vivo safety of MBZM-N-IBT observed in toxicity studies. However, it is necessary to closely evaluate its effect on renal and hepatic function for long-term therapy. Although at this dose, it does not affect motor coordination, locomotion, and anxiety, some sedation is observed. Hence, it is necessary to closely monitor its effect on the brain. Further investigation into mutagenicity and teratogenicity is also required. Thus, the present investigation can encourage additional studies to increase the scope for translational application of MBZM-N-IBT.
Acknowledgement
This work was supported by the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India.
Conflict of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding Sources
This work was supported by the Grant BT/PR15750/MED/29/1015/2016 from the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India.
Ethical Approval
The experimental procedure was approved by the Institutional Animal Ethics Committee (IAEC) of School of Pharmaceutical Sciences, Siksha O Anusandhan (Deemed to be University), Bhubaneswar, India (Reg. no. 1171/PO/Re/S/08/ CPCSEA).
References
- Rougeron V, Sam IC, Caron M, Nkoghe D, Leroy E, Roques P. Chikungunya, a paradigm of neglected tropical disease that emerged to be a new health global risk. J Clin Virol. 2015; 64: 144-152. http://dx.doi.org/10.1016/j.jcv.2014.08.032
CrossRef - Moizeis RN, Fernandes TA, Guedes PM, Pereira HW, Lanza DC, Azevedo JW, Galvao JM, Fernandes JV. Chikungunya fever: a threat to global public health. Pathog Glob Health. 2018; 112(4): 182-194. https://doi.org/10.1080/20477724.2018.1478777
CrossRef - Mathew AJ, Ganapati A, Kabeerdoss J, Nair A, Gupta N, Chebbi P, Mandal SK, Danda D. Chikungunya infection: a global public health menace. Curr Allergy Asthma Rep. 2017; 17: 1-9. https://doi.org/10.1007/s11882-017-0680-7
CrossRef - Franco EJ, Rodriquez JL, Pomeroy JJ, Hanrahan KC, Brown AN. The effectiveness of antiviral agents with broad-spectrum activity against chikungunya virus varies between host cell lines. Antivir Chem Chemother. 2018; 26: 2040206618807580. https://doi.org/10.1177/2040206618807580
CrossRef - Kaur P, Thiruchelvan M, Lee RC, Chen H, Chen KC, Ng ML, Chu JJ. Inhibition of chikungunya virus replication by harringtonine, a novel antiviral that suppresses viral protein expression. Antimicrob. Agents Chemother. 2013; 57(1): 155-167. https://doi.org/10.1128%2FAAC.01467-12
CrossRef - Subudhi BB, Chattopadhyay S, Mishra P, Kumar A. Current strategies for inhibition of chikungunya infection. Viruses. 2018; 10(5): 235. http://dx.doi.org/10.3390/v10050235
CrossRef - Staveness D, Abdelnabi R, Near KE, Nakagawa Y, Neyts J, Delang L, Leyssen P, Wender PA. Inhibition of Chikungunya virus-induced cell death by salicylate-derived bryostatin analogues provides additional evidence for a PKC-independent pathway. J Nat Prod. 2016; 79(4): 680-684. https://doi.org/10.1021%2Facs.jnatprod.5b01017
CrossRef - Bhakat S, Soliman ME. Chikungunya virus (CHIKV) inhibitors from natural sources: a medicinal chemistry perspective. J Nat Med. 2015; 69: 451-462. https://doi.org/10.1007/s11418–015-0910–z
CrossRef - Haese N, Powers J, Streblow DN. Small molecule inhibitors targeting chikungunya virus. Chikungunya Virus. 2020: 107-139. https://doi.org/10.1007/82_2020_195
CrossRef - Mishra P, Kumar A, Mamidi P, Kumar S, Basantray I, Saswat T, Das I, Nayak TK, Chattopadhyay S, Subudhi BB, Chattopadhyay S. Inhibition of chikungunya virus replication by 1-[(2-methylbenzimidazol-1-yl) methyl]-2-oxo-indolin-3-ylidene] amino] thiourea (MBZM-N-IBT). Sci Rep. 2016; 6(1): 20122. https://doi.org/10.1038/srep20122
CrossRef - Kumar A, De S, Moharana AK, Nayak TK, Saswat T, Datey A, Mamidi P, Mishra P, Subudhi BB, Chattopadhyay S. Inhibition of herpes simplex virus-1 infection by MBZM-N-IBT: in silico and in vitro studies. Virol J. 2021; 18(1): 103. https://doi.org/10.1186/s12985-021-01581-5
CrossRef - Chen L, Zhang B, Liu J, Fan Z, Weng Z, Geng P, Wang X, Lin G. Pharmacokinetics and bioavailability study of monocrotaline in mouse blood by ultra-performance liquid chromatography-tandem mass spectrometry. Biomed Res Int. 2018; 2018. https://doi.org/10.1155/2018/1578643
CrossRef - Sperling F. Introduction to toxicity evaluation session. Environ Health Perspect. 1979; 32: 259. https://doi.org/10.1289/ehp.7932259
CrossRef - Walum E. Acute oral toxicity. Environ Health Perspect. 1998; 106(2): 497-503. https://doi.org/10.1289%2Fehp.98106497
CrossRef - Akhila JS, Shyamjith D, Alwar MC. Acute toxicity studies and determination of median lethal dose. Curr Sci. 2007: 917-920. https://www.jstor.org/stable/24099255
- Moggs J, Moulin P, Pognan F, Brees D, Leonard M, Busch S, Cordier A, Heard DJ, Kammuller M, Merz M, Bouchard P. Investigative safety science as a competitive advantage for Pharma. Expert Opin Drug Metab Toxicol. 2012; 8(9): 1071-1082. https://doi.org/10.1517 /17425255.2012.693914
CrossRef - Weaver RJ, Valentin JP. Today’s challenges to de-risk and predict drug safety in human “mind-the-gap”. Toxicol Sci. 2019; 167(2): 307-321. https://doi.org/10.1093/toxsci/kfy270
CrossRef - Iliev I, Georgieva S, Sotirova Y, Andonova V. In silico study of the toxicity of hyperforin and its metabolites. Pharmacia. 2023; 70(3): 435-447. https://doi.org/10.3897/pharmacia.70.e107041
CrossRef - Bedi O, Krishan P. Investigations on acute oral toxicity studies of purpurin by application of OECD guideline 423 in rodents. Naunyn Schmiedebergs Arch. Pharmacol. 2020; 393: 565-571. https://doi.org/10.1007/s00210-019-01742-y
CrossRef - Jonsson M, Jestoi M, Nathanail AV, Kokkonen UM, Anttila M, Koivisto P, Karhunen P, Peltonen K. Application of OECD Guideline 423 in assessing the acute oral toxicity of moniliformin. Food Chem. Toxicol. 2013; 53: 27-32. https://doi.org/10.1016/j.fct.2012.11.023
CrossRef - Arpornchayanon W, Subhawa S, Jaijoy K, Lertprasertsuk N, Soonthornchareonnon N, Sireeratawong S. Safety of the Oral Triphala Recipe from Acute and Chronic Toxicity Tests in Sprague-Dawley Rats. Toxics. 2022; 10(9): 514. https://doi.org/10.3390/toxics10090514
CrossRef - Guideline IH. Validation of analytical procedures: text and methodology. Q2 (R1). 2005; 1(20): 05. https://database.ich.org/sites/ default/files/Q2%28R1%29%20Guideline.pdf
- Sabale V, Jiwankar M, Sabale P. Bioanalytical method development, validation and quantification of flutamide in spiked rat plasma by using high-performance liquid chromatography. Future J. Pharm. Sci. 2023; 9(1): 1-7. https://doi.org/10.1186/s43094-023-00528-7
CrossRef - Hegde AR, Padya BS, Soman S, Mutalik S. A simple, precise, and sensitive HPLC method for quantification of letrozole in rat plasma: development, validation, and preclinical pharmacokinetics. J. Anal. Sci. Technol. 2021; 12(1): 1-8. https://doi.org/10.1186/s40543-021-00276-4
CrossRef - Li YF, Zhang XQ, Hu WY, Li Z, Liu PX, Zhang ZQ. Rapid screening of drug-protein binding using high-performance affinity chromatography with columns containing immobilized human serum albumin. J Anal Methods Chem. 2013; 2013. https://doi.org/10.1155/2013/439039
CrossRef - Marin DE, Taranu I. Using In Silico Approach for Metabolomic and Toxicity Prediction of Alternariol. Toxins. 2023; 15(7): 421. https://doi.org/10.3390/toxins15070421
CrossRef - Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong J, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick CM, Ray JE. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011; 50: 81-98. https://doi.org/10.2165/11534750-000000000-00000
CrossRef - de C Bergamaschi C, Berto LA, Venancio PC, Cogo K, Franz-Montan M, Motta RH, Santamaria MP, Groppo FC. Concentrations of metronidazole in human plasma and saliva after tablet or gel administration. J. Pharm. Pharmacol.2014; 66(1): 40-47. https://doi.org/10.1111/jphp.12161
CrossRef - Roberts CJ. Clinical pharmacokinetics of ranitidine. Clin. Pharmacokinet. 1984; 9(3): 211-221. https://doi.org/10.2165/00003088-198409030-00003
CrossRef - Gazzard BG, Ford-Hutchinson AW, Smith MJ, Williams R. The binding of paracetamol to plasma proteins of man and pig. J. Pharm. Pharmacol. 1973; 25(12): 964. https://doi.org/10.1111/j.2042-7158.1973.tb09987.x
CrossRef - Patsalos PN, Patsalos PN. Antineoplastic Agents. Antiepileptic Drug Interactions: A Clinical Guide. 2016: 253-262.https://doi.org/10.1007/978-3-319-32909-3
CrossRef - Barre J. Pharmacokinetic properties of cefixime. Presse Medicale (Paris, France: 1983). 1989; 18(32): 1578-1582. https://pubmed.ncbi.nlm.nih.gov/2530536/
- Cook CS, Schoenhard GL, Karim A. Effect of salicylic acid on the plasma protein binding and pharmacokinetics of misoprostol acid. J Pharm Sci. 1994; 83(6): 883-886. https://doi.org/10.1002/jps.2600830625
CrossRef - Perona A, Ros MP, Mills A, Morreale A, Gago F. Distinct binding of cetirizine enantiomers to human serum albumin and the human histamine receptor h1. J. Comput. Aided Mol. Des. 2020; 34(10): 1045-1062. https://doi.org/10.1007/s10822-020-00328-8
CrossRef - Macek J, Klíma J, Ptacek P. Rapid determination of omeprazole in human plasma by protein precipitation and liquid chromatography–tandem mass spectrometry. J. Chromatogr. B. 2007; 852(1-2): 282-287. https://doi.org/10.1016/j.jchromb.2007.01.026
CrossRef - Moeinpour F, Mohseni-Shahri FS, Malaekeh-Nikouei B, Nassirli H. Investigation into the interaction of losartan with human serum albumin and glycated human serum albumin by spectroscopic and molecular dynamics simulation techniques: A comparison study. Chem. Biol. Interact. 2016; 257: 4-13. https://doi.org/10.1016/j.cbi.2016.07.025
CrossRef - De S, Ghosh S, Keshry SS, Mahish C, Mohapatra C, Guru A, Mamidi P, Datey A, Pani SS, Vasudevan D, Beuria TK. MBZM-N-IBT, a novel small molecule, restricts chikungunya virus infection by targeting nsP2 protease activity in vitro, in vivo, and ex vivo. Antimicrob Agents Chemother. 2022; 66(7): e00463-22. https://doi.org/10.1128/aac.00463-22
CrossRef - Smith DA, Beaumont K, Maurer TS, Di L. Relevance of half-life in drug design: Miniperspective. J. Med. Chem. 2017; 61(10): 4273-4282. https://doi.org/10.1021/acs.jmedchem.7b00969
CrossRef - Mombelli E, Pandard P. Evaluation of the OECD QSAR toolbox automatic workflow for the prediction of the acute toxicity of organic chemicals to fathead minnow. Regul. Toxicol. Pharmacol. 2021; 122: 104893. https://doi.org/10.1016/j.yrtph.2021.104893
CrossRef

