
Arwa Mustafa1 , Fathelrahman Mahdi Hassan2
, Fathelrahman Mahdi Hassan2![]() , Abdelgadir Ahmed1, Mawadah Yousif3, Sahar G Elbager4 and Ahmed Gaffer5
, Abdelgadir Ahmed1, Mawadah Yousif3, Sahar G Elbager4 and Ahmed Gaffer5
1Faculty of medical laboratory sciences, Omdurman Islamic University, Sudan.
2Department of hematology and immunohematology, College of medical laboratory science, Sudan University of Science and Technology, Khartoum, Sudan.
3Institute of Endemic Diseases, University of Khartoum, Sudan.
4Faculty of medical laboratory sciences, University of medical sciences and Technology, Khartoum, Sudan.
5Faculty of medical laboratory sciences, Alzaeim alazhari university Khartoum, Sudan.
Corresponding Author E-mail: ahmedkodab2009@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/2881
Abstract
Background: BCL11A is associated with HbF in many populations with different variants of polymorphisms, our study aimed to estimate the prevalence Bcl11a polymorphisms and its association with HbF in Sudanese sickle cell patients Materials and methods: This study was done in Jafar Ibn Ouf Children's Hospital from March to August 2019 DNA was extracted using the phenol-chloroform technique, the Bcl11A was processed for (rs10184550), and (rs7599488) Sanger sequencing was used to detect polymorphisms after the purification of DNA. Results: A total of 21 were successfully sequenced, 21 were screened for SNPs (rs10184550) while 7 patients were screened for both BCL11A (rs10184550) and (rs7599488) polymorphisms. Single nucleotide polymorphisms (SNPs) (rs10184550) 16 (76.2%) were males and 5 (23.8%) were females with a mean age of 6.6± 2.9 years (range 2-12 years), The mean HbF level was 16.9±6.6%. For rs10184550 polymorphisms, out of 21 patients, 11 /21 (52%) patients had the “A” allele, and 10/21 (48%) patients had the “G” allele. For, rs7599488, the “T” allele was detected in 1/7 (14%) patients. In comparison, the “C” allele was detected in 6/7 (86%) patients. Out of 7 patients who screened for BCL11A (rs10184550) and (rs7599488) polymorphisms, 6 patients have rs10184550 “G” allele and rs7599488 “C” allele, while one patient had rs10184550 “A” allele and rs7599488 “C” allele. Furthermore, patients with allele “A” rs10184550 have a significantly higher mean HbF level than patients with the “G” allele (20.1± 6.2 vs 13.43± 5.5, p =0.01 Conclusion: Bcl11 polymorphism is associated with high haemoglobin F in Sudanese sickle cell patients.
Keywords
Anaemia; polymorphism; Sickle cell disease
Download this article as:| Copy the following to cite this article: Mustafa A, Gmeel F, Ahmed A, Yousif M, Elbager S. G, Gaffer A. Genetic Polymorphisms at BcL11A Sites rs10184550 and rs7599488 in Sudanese Sickle Cell Patients. Biomed Pharmacol J 2024;17(1). |
| Copy the following to cite this URL: Mustafa A, Gmeel F, Ahmed A, Yousif M, Elbager S. G, Gaffer A. Genetic Polymorphisms at BcL11A Sites rs10184550 and rs7599488 in Sudanese Sickle Cell Patients. Biomed Pharmacol J 2024;17(1). Available from: https://bit.ly/49aZsOY |
Introduction
In 1950 the first case of HbS was reported 1, later, several studies were conducted and showed that the sickle cell gene frequency differed from region to region. In central Sudan(Khartoum state) among 623 patients attending different clinics 5.1 % with HbAS and 0.8 % SS2, in the Blue Nile the prevalence ranges from 0-5% in addition to the rate of 16% in migrant African tribes from western Sudan and west Africa the north Sudan the sickle cell gene was not detected in Shaigia and Mansir tribes and reported as zero3. In Eastern Sudan, the study was conducted in Hussa (261)and Massalet (285) with a total of 546 study participants, the prevalence of heterozygous sickle cell(AS) was 35% in Hausa and 24% In Masaleet whereas HbSS was 6%and 5% in Hausa and Masaleet respectively4. B/cell lymphoma leukaemia (Bcl11A) which involves B-cell progenitors production is highly expression in many cells, it plays a crucial role in the fusion γ to B-globin which fuses the fetal haemoglobin(HbF) to adult type (HbA)5,6,7. However, down-regulation of these genes in adult erythroid precursors results in the induction of HbF. A study revealed that SNP in Bcl11A(rs11886868) strongly correlate with HbF in sickle cell disease8.
Three studies were done in Sudan to study HbF silencing factors one study demonstrated BCL11A in sickle cell patients, they concluded that BCL11A was associated with HbF level and the SNP (rs11886868 and rs766432) at the above region are associated with HbF level, the severity of the disease was not assessed9. Another study concluded that the absence of XmnI was found in 3% of SS patients and was associated with HbF or disease severity10. The last study demonstrated the same locus XmnI they summarized that patients with XmnI in both sites had higher HbF than patients with one site XmnI11. Here we studied the BCL11A (rs10184550) and (rs7599488) and its association with HbF level in Sudanese children affected by SCD.
Materials and methods
This is part of a cross-sectional study done in Jafar Ibn Ouf Children’s Hospital from March to August 2019 which includes 25 patients aged 3-12 years old who were selected for the sequencing of different SNPs in Bcl11A locus, sickle cell patients with high Hb F were selected for this study, while patients with crises, transfused within the last three months, and with other hemoglobinopathies were excluded from this study. Data were collected with a pre-coded questionnaire prepared for this study, the sample was drowned with a 5ml syringe under the aseptic condition in an EDTA tube, and Hb was separated by electrophoresis to confirm the HbS (CAPILLARYS 2 FLEX-PIERCING), DNA was extracted using previously published methods, the Bcl11A was processed with following primers for (rs10184550), and (rs7599488)
Forward: S2:CCACCAGAAGTCCTGGAAA,
Reverse: GTGTGATGGCAGAGCTCAAA,
Forward: S3:GCCAGAGCACACTTGAATGA
Reverse: TCTGAGGGCCTTCGAACTTA.
PCR was performed with following concentrations (PCR Master mix =6µ, Primer Forward =1µ, Primer reveres= 1µ, H20 = 17µ ,DNA = 5µ ,Total = 30µ). The amplification was carried out by an initial denaturation stage at 95˚C for 5 min, then 30 cycles of 94˚C for 30 seconds and 60˚C for 30 seconds, 72˚C for 1 min, and a final extension for 5 min at 72˚C. The PCR products were shown in standards electrophoresis in which the sample was separated according to molecular weight (S2 S3. The result of gels was detected by UV light with S2=403bp S3=400bp figure as in figures (1 and 2) and sequencing was done to detect the polymorphisms in specific bases.
Ethical clearance
Ethical approval was obtained from the Omdurman Islamic University faculty of medical laboratory sciences ID (#0419), and parents of the children were assigned to get consent after they were informed about the aim of the study the data was taken in the questionnaire after the approval of all children parents.
Data analysis
The data was processed and analyzed using a statistical package for social sciences (SPSS) version 24. An Independent T-test was used to calculate P.value and P.value <0.005 considered significant. Sanger sequencing was employed as a reference method, to verify the obtained genotypes. For this purpose, the samples were subjected to sequencing. The results of Sanger sequencing were detected using BioEdit software version 7.0.5.3. The reference sequence of the BCL11A gene (NG_01196) was retrieved from NCBI (https://www.ncbi.nlm.nih.gov/). The BioEdit software was used for multiple sequence alignment and chromatogram sequence visualization
Results
A total of 25 children with sickle cell anaemia (HbSS) were enrolled in this study and 4 did not meet the criteria for sequencing, the remaining 21 were successfully sequenced. 21 were screened for SNPs (rs10184550) while 7 patients were screened for both BCL11A (rs10184550) and (rs7599488) polymorphisms. Positive gel with Bcl11A was sent for sequencing as in Figures 1 and 2. For SNPs (rs10184550) 16 (76.2%) were males and 5 (23.8%) were females with a mean age of 6.6± 2.9 years (range 2-12 years), the mean HbF level was 16.9±6.6%. For rs10184550 polymorphisms, out of 21 patients, 11 /21 (52%) patients have the “A” allele, and 10/21 (48%) patients have the “G” allele (figure .3). For, rs7599488, the “T” allele was detected in 1/7 (14%) patients and the “C” allele was detected in 6/7 (86%) patients (fig.4). Out of 7 patients who screened for BCL11A (rs10184550) and (rs7599488) polymorphisms, 6 patients have rs10184550 “G” allele and rs7599488 “C” allele, while one patient had rs10184550 “A” allele and rs7599488 “C” allele. Furthermore, patients with allele “A” rs10184550 have a significantly higher mean of HbF levels than patients with the “G” allele (20.1± 6.2 vs 13.43± 5.5, p =0.01). Table 2.
 |
Figure 1: PCR product on gel 400bp(S3) lane 1 DNA Loader, lane 2 negative control lane (3-9) samples from |
 |
Figure 2: PCR product S2 (403bp) Lane1 negative control lane 2 loader lane 3-9 samples |
Table 1: Demographics and hematological data of SCA patients.
|
Variables |
Patients (n=21) |
|
Gender M, n(%) F, n(%) |
16 (76.2%) 5 (23.8%) |
|
Age (Mean± STD) years |
6.6± 2.9 y |
|
Hb A (Mean± STD) g/dl |
8.9 ±0.8 |
|
Hb F (Mean± STD) % |
16.9±6.6 |
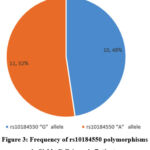 |
Figure 3: Frequency of rs10184550 polymorphisms in Sickle Cell Anemia Patients |
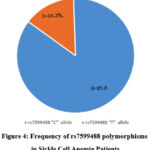 |
Figure 4: Frequency of rs7599488 polymorphisms in Sickle Cell Anemia Patients |
Table 2: Mean of HbF levels in SCA patients according to rs10184550 polymorphism
|
rs10184550 |
GG (n=10) |
GA (n=11) |
p-value |
|
Hb F |
13.43± 5.5 |
20.1± 6.2 |
0.01 |
Discussion
BCL11A is well documented to be associated with Hbf in many populations, Considering SNP rs 7599488 it is consistent with African alleles frequency which the C alleles were common (0.678) and near to our study in which the frequency of the C allele is 0.857 with a slight increase in our study1. For the rs10184550 our result is consistent with the European allele frequency and in Latin America, the wild-type allele frequencies were 0.444 and 0.417 and our result is 0.47613. A study of mapping of loci associated with HbF in African Americans with sickle cell anaemia demonstrated that rs7599488 as BCL11A loci is associated with HbF in which T alleles predominated. In addition same study, it is concluded that the allelic variance it antagonistic effect with haplotype to affect the HbF which is affected by haplotypes and SNP14, however here we didn’t determine the haplotypes which are largely different from the other countries, here the comparison is difficult due to the low numbers (only seven samples). Furthermore, this allelic frequency at rs 7599488 was confirmed by another study in different populations of African American, western and eastern Saudi Arabs, and Indian populations in which the frequency of the T/C allele were 0.30, 0.42, 0.43, and 0.68 respectively15. In the Mexico population, rs 7599488 failed to be associated with HbF in a small sample of homozygous sickle cell samples16. Bcl11a(rs10184550) is recently identified as having a therapeutic role in the epileptic patient17 to the central role in the HbF silencing factors. A study done in Columbia found that the rs rs10184550 is significantly associated with high HbF in the patient under hydroxy urea, with the G allele this is consistent with our result in which G18, the same finding in the Chinese study which found rs10184550 is associated with high HbF in beta-thalassemia and ameliorating the diseases19.
Conclusion
In conclusion, the bcl11a polymorphisms are associated with HbF in our population the limitations of this study are the small sample size and failure to determine the haplotypes, in addition to, the Sudanese reservoir of Cameron/Cameron haplotypes and others haplotypes. Further study with a large sample size including controls with haplotypes of sickle cell is required.
Acknowledgement
None
Conflict of Interest
There is no conflict of interest.
Funding Sources
There are no funding sources
References
- El-Hazmi MA, Al-Hazmi AM WA. Sickle cell disease in Middle East Arab countries. Indian J Med Res. 2011;134:597–610.
CrossRef - Elderdery AY, Mohamed BA, Cooper AJ, Knight G MJ. Sickle cell disease in Middle East Arab countries. J Med Lab Diagnosis August. 2011;2:31–7.
- Kempińska-Podhorodecka A. et al. Sickle Cell Anemia-Associated Beta-Globin Mutation in Shagia and Manasir Tribes from Sudan. Pol J Environ Stud. 2011;20(6):1525–30.
- Salih NA, Hussain AA, Almugtaba IA, Elzein AM et al. Loss of balancing selection in the betaS globin locus. BMC Med Genet. 2010;3:21.
CrossRef - Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science (80- ). 2008;322(5909).
CrossRef - Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6).
CrossRef - Lettre G et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A. 2008;105:11869–11874.
CrossRef - Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science (80- ). 2013;342(6155).
CrossRef - Shireen ZN, Elshazali WA HK. Association of BCL11A Genetic Polymorphisms with Fetal haemoglobin Level in Sudanese Patients with Sickle Cell Anaemia. J Genom Gene Study. 2019;2(1).
- Rja.AO NM. The association of the -158 XmnI γG globin polymorphism with HbF level in sickle cell anemia Sudanese Patients. J Biosci Appl Res. 2022;8(1).
- Osman T, Ahmed K, Alshazaly A, Altag A, Abdalla A, Eldour A et al. Association of XmnI Polymorphism with Fetal Hemoglobin Level in Sudanese Patients with Sickle Cell Disease. Int J Contemp Med. 2021;9(1):31–4.
- ALFA: Allele Frequency [Internet]. 2022. Available from: https://www.ncbi.nlm.nih.gov/snp/rs7599488#frequency_tab
- dbSNP Short Genetic Variations. 2022; Available from: https://www.ncbi.nlm.nih.gov/snp/rs10184550#frequency_tab
- Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN LG. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42(12):1049–51.
CrossRef - Sebastiani P, Farrell JJ, Alsultan A, Wang S, Edward HL, Shappell H, et al. BCL11A enhancer haplotypes and fetal hemoglobin in sickle cell anemia. Blood Cells, Mol Dis. 2015;54(3).
CrossRef - Rizo-de la Torre LC, Borrayo-López FJ, Perea-Díaz FJ, Aquino E, Venegas M, Hernández-Carbajal C, et al. Fetal hemoglobin regulating genetic variants identified in homozygous (HbSS) and heterozygous (HbSA) subjects from South Mexico. J Trop Pediatr. 2022;68(5).
CrossRef - Wang S, Cai X, Liu S, Zhou Q, Wang T, Du S, et al. A novel BCL11A polymorphism influences gene expression, therapeutic response and epilepsy risk: A multicenter study. Front Mol Neurosci. 2022;15.
CrossRef - Green NS, Ender KL, Pashankar F, Driscoll C, Giardina PJ, Mullen CA, et al. Candidate Sequence Variants and Fetal Hemoglobin in Children with Sickle Cell Disease Treated with Hydroxyurea. PLoS One. 2013;8(2).
CrossRef - Sedgewick AE, Timofeev N, Sebastiani P, So JCC, Ma ESK, Chan LC, et al. BCL11A is a major HbF quantitative trait locus in three different populations with β-hemoglobinopathies. Blood Cells, Mol Dis. 2008;41(3).
CrossRef

