
Manuscript accepted on :17-02-2023
Published online on: 13-04-2023
Plagiarism Check: Yes
Reviewed by: Dr. Liudmila Spirina
Second Review by: Dr. S Shahi
Final Approval by: Dr. H Fai Poon
Mamatha Kunder1* , A.V. Moideen Kutty2 and V. Lakshmaiah3
, A.V. Moideen Kutty2 and V. Lakshmaiah3
1Department of Biochemistry, Yenepoya School of Allied Health Sciences, Yenepoya (Deemed to be) University, Mangalore, Karnataka, India.
2Yenepoya (Deemed to be) University, Mangalore, Karnataka, India.
3Department of Medicine, Sri Devaraj Urs Medical College, Sri Devaraj Urs Academy of Higher Education and Research, Kolar, Karnataka, India.
Corresponding Author E-mail: mamathayogesh79@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/2669
Abstract
Neutrophils are the first to infiltrate ischemic brain regions causing the release of Neutrophil Elastase (NE), a pro-inflammatory proteinase. The activity of NE is well regulated by endogenous inhibitors alpha1-antitrypsin (α1-AT) and alpha2-macroglobulin (α2-MG). The physiological balance of elastase and anti-elastase factors is essential to maintain the normal integrity of tissues and an imbalance has been implicated in the pathogenesis of several acute and chronic inflammatory diseases. The present study was designed to determine the plasma levels of NE, α1-AT, α2-MG, and NE–α1-AT complex to evaluate their role in inflammatory processes of ischemic stroke. The effect of homocysteine on the release of elastase from neutrophils was also studied. The study involved a total of 100 subjects (controls =60 and patients=40). Significantly higher mean elastase activity and lower α1-AT levels were observed in ischemic stroke patients than in controls. NE- α1-AT complex and α2-MG levels were significantly increased in the patient group. The in vitro study indicated homocysteine induced release of elastase from neutrophils. In conclusion, homeostasis of NE and its endogenous inhibitors is deranged in patients suggestive of their role in the pathogenesis of ischemic stroke through exacerbating inflammatory and coagulation processes.
Keywords
α1-Antitrypsin; α2-Macroglobulin; Acute Ischemic Stroke; Homocysteine; Neutrophil Elastase
Download this article as:| Copy the following to cite this article: Kunder M, Kutty A. V. M, Lakshmaiah V. Derangement in Homeostasis of Neutrophil Elastase and its Inhibitory Systems in Ischemic Stroke Patients. Biomed Pharmacol J 2023;16(2). |
| Copy the following to cite this URL: Kunder M, Kutty A. V. M, Lakshmaiah V. Derangement in Homeostasis of Neutrophil Elastase and its Inhibitory Systems in Ischemic Stroke Patients. Biomed Pharmacol J 2023;16(2). Available from: https://bit.ly/3UzPe4g |
Introduction
Inflammation plays an important role in the pathogenic progression of ischemic stroke. Acute ischemic stroke accounts for about 85% of all cases due to the sudden decrease or loss of blood circulation to an area of the brain, resulting in a corresponding loss of neurological function. The brain responds to ischemic injury by rapidly initiating the inflammatory cascade which is characterized by rapid activation of microglial cells, production of proinflammatory mediators, and infiltration of inflammatory cells into the ischemic brain tissue 1, 2, 3.
Among the various types of inflammatory cells, neutrophils are the first to infiltrate ischemic brain regions which occur as early as thirty minutes after ischemia and reperfusion with the degree of accumulation correlated with the extent of brain infarct and clinical outcomes4, 5. Once activated, the infiltrating neutrophils release a variety of cytotoxic agents including proteolytic enzymes, free radicals, and cytokines which further amplify the inflammatory responses in the brain, with damaging effects on the Blood-Brain Barrier (BBB)6, 7. Disruption of BBB further potentiates brain tissue injury and contributes to secondary ischemic brain damage3. Experimental studies have shown neutrophil elastase (NE) to have a major role in the breakdown of BBB and pharmacological inhibition of NE significantly reduced infarct volume, BBB disruption, and leukocyte-endothelial adherence 8, 9, 10.
The activity of NE released to the extracellular region is well regulated and controlled by a number of endogenous protease inhibitors such as alpha1-antitrypsin (α1-AT) and alpha2-macroglobulin (α2-MG) by irreversibly inactivating the free enzyme by forming 1:1 complex with it11. Adequate levels of these inhibitors are shown to have effects in mitigating the severity of inflammatory processes. Studies have shown significant alterations in the levels of these endogenous inhibitors in stroke patients12, 13, 14.
The role of neutrophil elastase and its endogenous inhibitors in the inflammatory process and their consequential role in the pathophysiological processes of stroke have been previously studied13, 14, 15, 16. However, a comprehensive evaluation of the levels of NE, its endogenous inhibitors α1-AT and α2-MG, and NE- α1-AT complex has not been conducted in stroke patients. This study was therefore designed to determine the plasma levels of these molecules in ischemic stroke patients.
Further, many experimental and clinical studies have demonstrated hyperhomocysteinemia as an independent risk factor for the development and progression of vascular diseases such as stroke17, 18, 19. Since NE is known to play a key role in the inflammatory processes of stroke, an in vitro experimental setup was designed to study the effect of homocysteine on the release of elastase from neutrophils.
Materials and Methods
Patient selection
A total of 100 subjects were included in the study and among them, 60 were healthy individuals and 40 were first-ever ischemic stroke patients admitted at R. L. Jalappa Hospital and Research Centre Kolar, Karnataka. Informed written consent was obtained from every individual or the nearest kin of every patient. The study protocol was approved by the Institutional Ethical Committee of Sri Devaraj Urs Medical College, a constituent college of Sri Devaraj Urs Academy of Higher Education and Research, Kolar, Karnataka and the study complied with the Helsinki Declaration.
Ischemic stroke was confirmed by imaging studies (Magnetic Resonance Imaging or Computed Tomography). Patients with non-ischemic stroke, coronary heart disease, diabetes mellitus, malignancy, acute and chronic infections, chronic obstructive lung disease, liver disorders, and acute renal failure as well as a history of smoking were excluded from the study.
Sample collection
Blood samples were obtained from all patients within 48 hours of admission and before antithrombotic administration to avoid potential interferences with the estimation of study molecules. 6ml of Venous blood was collected from an antecubital vein into tubes containing sodium heparin (for elastase, α1-AT and α2-MG and NE–α1-AT complex estimations) and EDTA (for HbA1c and Complete Blood count). For investigations such as CRP, renal function tests and liver function tests blood was collected in tubes without anticoagulant. The blood samples were centrifuged for 15 minutes within 2h of collection. Routine parameters were analyzed immediately and aliquots were stored at -80ºC for estimation of NE and its endogenous inhibitors. Samples were thawed at room temperature and centrifuged before analysis.
Chemicals and Reagents
All reagents used for the study were of analytical grade. Succinyl tri- L-alanyl-p-nitroanilide (STANA, Cat.No. S4760) and DL-Homocysteine (Cat.No. 44925) were procured from Sigma, USA. α1-AT and α2-MG ELISA kits were purchased from Immunology Consultants Laboratory, Inc, USA, and NE–α1-AT complex kit from Calbiochem, USA. Fetal Bovine Serum (2% FBS, Cat. No. 10270106) and RPMI-1640 media (Cat. No. 31800022) were purchased from Invitrogen. Ficoll-paque Plus was obtained from GE Healthcare (Cat.No. 17-1440-02). Dextran (20%) was obtained from Himedia (Cat.No. RM 4187) and tissue culture plates (96-well) were procured from Genetix.
Methods
Routine biochemical estimations were carried out by standard methods using Vitros 250 Dry chemistry analyzer (Ortho Clinical Diagnostics, USA). HbA1c was analyzed by HPLC method using Bio-Rad D-10. Complete Blood Count was performed by Beckman-Coulter’s automatic blood cell counter. Serum CRP estimation was done by rapid latex slide tests. Plasma elastase was estimated using Succinyl tri- L-alanyl-p-nitroanilide as substrate at 410nm as per the procedure described by Beith J et al20. Plasma α1-AT, α2-MG, and NE–α1-AT complex levels were quantified by ELISA as per the manufacturer’s instructions.
Isolation and In vitro culture of neutrophils
Neutrophils were isolated from the peripheral blood of healthy volunteers by Ficoll- dextran density gradient method as described by Joshi et al21. Cell purity was assessed by Giemsa staining and cell viability was determined by the trypan blue dye exclusion method. Purified neutrophils were re-suspended in a culture medium of RPMI with 2% FBS.
Neutrophils (2.5×106) were seeded on 96-well tissue culture plates and incubated with increasing concentrations of homocysteine (5, 11, 25, 50, 200µM/L) in a final volume of 100µl per well at 37oC in a CO2 incubator for 2hours. Three independent experiments were carried out for each concentration and incubation time. The culture supernatants from each set were collected after an incubation period of 30 minutes and were immediately assayed for elastase activity using STANA as substrate.
Statistical Analyses
The data were analyzed by SPSS software version 22(licensed version 22, IBM Corp, USA) for statistical significance. The results are expressed as mean ± SD. All variables were checked for normal distribution by Shapiro–Wilk test. For statistical differences between the groups’ Student’s unpaired t-test was used. Graph-Pad Prism 5.0 Software was used to analyze the in vitro study data. ANOVA followed by post hoc Bonferroni’s tests was used to calculate the statistically significant differences between the experimental groups. P values <0.05 was considered statistically significant and <0.001 as highly significant.
Results and Discussion
The baseline characteristics of study groups are depicted in Table 1. The mean age was relatively higher in stroke patients (65.60+9.36) compared to the control group (50.47±7.99). Majority were males in the study groups. The systolic and diastolic blood pressure was significantly higher in stroke patients compared to control subjects. The data on serum CRP presented significantly higher levels in stroke patients reflecting systemic inflammation in them (25.35+5.62).
Table 1: Basic characteristics of Study groups
| Variables | Control (n=60) | Stroke (n=40) |
| Age (years)* | 50.47±7.99 | 65.60+9.36 |
| Gender(male/female) | 35/25 | 32/8 |
| Blood Pressure (mmHg)*
Systolic Diastolic |
120.28+8.70 80.20+5.88 |
200.5+36.08 114.0+14.82 |
| Serum CRP (ug/ml)* | 0 | 25.35+5.62 |
Values are expressed as Mean+SD. *P value is highly significant.
The results on mean plasma elastase activity showed significantly higher levels in patients with ischemic stroke (0.51+0.1) than in the control group (0.35±0.20) (Table 2). NE released from activated neutrophils contributes to the inflammatory response and is shown to play a key role in the pathogenesis and progression of stroke9, 22, 23. Significantly elevated levels of elastase activity were observed in stroke patients than in the control group in the current study thus confirming the role of NE in progressive inflammation as reported earlier15, 24.
Alpha1-antitrypsin is a potent anti-inflammatory, anti-apoptotic, and cytoprotective biomolecule that has been tested by various researchers for its beneficial role in stroke. Being a primary inhibitor of NE activity, an adequate concentration of this inhibitor is critical for protease-anti-protease homeostasis. A reduction in the levels of α1-AT thus could lead to NE exacerbating uncontrolled and injurious effects. In the present study, significantly reduced levels of α1-AT were observed in patients with ischemic stroke (87.63+27.69) compared to controls (122.95±25.71) (Table 2). It is well known that patients suffering from stroke exhibit an increase in oxidative stress and generate free radicals25. Free radicals have been shown to cause oxidative damage to α1-AT and this could be one of the reasons for the decrease in the levels of this inhibitor26. Moreover, there was a significant increase in the levels of NE- α1-AT complex in stroke patients (346.2+57.89) as compared to controls (215.83±13.61) (Table 2). This could be another reason for the decrease in α1-AT, as it forms a complex with NE to counteract the effects of NE. Furthermore, studies have found that a common variant of the α1-AT gene (M1 variant) is a risk factor for ischemic stroke, implying that the α1-AT gene plays a role in the pathogenesis of stroke27, 28.
When the plasma levels of α2-MG were compared, stroke patients had significantly higher levels (247.33+63.41) when compared to the control group (208.87±31.16) (Table 2). α2-MG is a broad-spectrum protease inhibitor that enhances pro-coagulant properties via the neutralization of plasmin, plasminogen activators, and metalloproteinases. The pro-coagulant properties of elevated α2-MG levels could independently contribute to intravascular coagulation adding to the severity of complications of stroke. Studies have associated elevated levels of α2-MG with high-grade white matter lesions implicating this inhibitor in the pathophysiology of acute ischemic stroke 13, 14.
Table 2: Plasma levels of elastase activity, α1-AT, α2-MG and NE-α1-ATcomplex in the study groups
| Parameters | Control (n=60) | Stroke (n=40) | p value
|
| Plasma Elastase activity (U/ml) | 0.35±0.20 | 0.51+0.1 | *<0.001
|
| Plasma α1-AT (mg/dl) | 122.95±25.71 | 87.63+27.69 | *<0.001
|
| Plasma α2-MG (mg/dl) | 208.87±31.16 | 247.33+63.41 | *<0.001
|
| Plasma NE-α1-ATcomplex (ng/ml) | 215.83±13.61 | 346.2+57.89 | *<0.001
|
Values are expressed as Mean+SD. *statistically highly significant.
Effect of Homocysteine on elastase release from neutrophils
In addition to the risk factors such as hypertension, diabetes, and smoking, studies have found that homocysteine is not only a risk factor for, but also correlated with, stroke severity, bad prognosis, and stroke recurrence29,30. Studies have found that hyperhomocysteinemia induces an elastolytic process in the arterial wall, by inducing synthesis and secretion of serine elastase causing the release of chemotactic elastin peptides. These peptides in turn induce vascular smooth muscle cell proliferation and migration into the subendothelium, leading to neointima formation and progressive vascular occlusion 31. In the present in vitro study, it was found that the release of elastase from the cultured neutrophils increased with concentrations of homocysteine and time (Figure 1). After reaching a maximum activity at 120 minutes of incubation further increase in homocysteine concentrations led to a decrease in elastase release which could be due to either limiting factors or the death of cells. The findings support that hyperhomocysteinemia could be an inducer of the release of elastase from neutrophils and might contribute to the onset of stroke mediated through an inflammatory response.
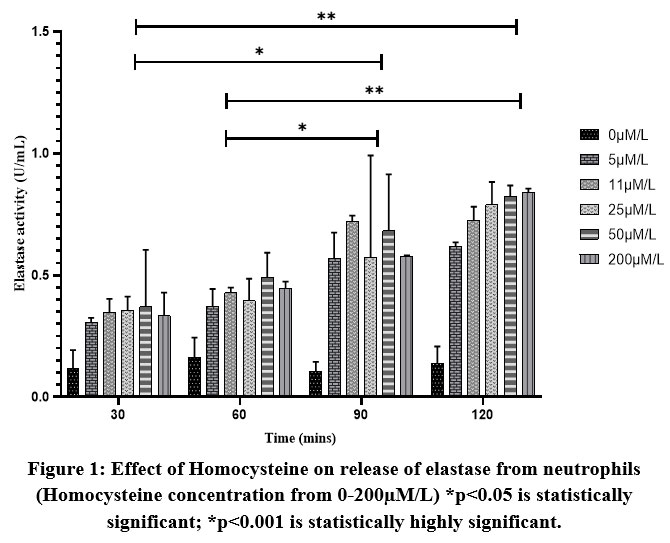 |
Figure 1: Effect of Homocysteine on release of elastase from neutrophils (Homocysteine concentration from 0-200µM/L) *p<0.05 is statistically significant; |
Conclusion
This study indicates that there is a disturbance in the homeostasis of neutrophil elastase and its inhibitory system in ischemic stroke patients. The increased levels of α2-MG and the potentiating role of homocysteine in inducing the release of elastase from neutrophils in vitro are suggestive of their roles in augmenting coagulation and inflammation, the leading events in ischemic stroke.
Acknowledgment
The authors would like to express their gratitude to Sri Devaraj Urs Academy of Higher Education and Research, Kolar, for carrying out this work.
Conflict of Interest
The authors have no conflicts of interest to declare.
Funding Sources
There are no funding sources
References
- Jin R, Yang G and Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J. Leukoc. Biol., 2010;87(5):779-89.
CrossRef - Lakhan S. E, Kirchgessner A and Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J. Transl. Med., 2009; 7:97.
CrossRef - Wang Q, Tang X. N and Yenari M. A. The Inflammatory Response in Stroke. J. Neuroimmunol., 2007; 184:53–68.
CrossRef - Yilmaz G and Granger D. N. Cell adhesion molecules and ischemic stroke. Neurol. Res., 2008; 30:783–93.
CrossRef - Kriz J. Inflammation in ischemic brain injury: timing is important. Crit. Rev. Neurobiol., 2006; 18:145–57.
CrossRef - Jickling G. C, Liu D, Ander B. P, Stamova B, Zhan X and Sharp F. R. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J. Cereb Blood Flow Metab., 2015; 35 (6):888-01.
CrossRef - Segel G. B, Halterman M. W and Lichtman M. A. The paradox of the neutrophil’s role in tissue injury. J. Leukoc. Biol., 2011; 89: 359–72.
CrossRef - Ikegame Y, Yamashita K, Hayashi S, Yoshimura S, Nakashima S and Iwama T. Neutrophil elastase inhibitor prevents ischemic brain damage via reduction of vasogenic edema. Hypertens. Res., 2010; 33:703–7.
CrossRef - Stowe A. M, Adair-Kirk T. L, Gonzales E. R, Perez R. S, Shah A. R, Park T. S., et al. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiology of disease., 2009; 35(1): 82-90.
CrossRef - Akira S, Yoshihisa K, Yasuhiko I, Kazunao K, Yasuhiro S and Kazuo U. Neutrophil elastase inhibition reduces cerebral ischemic damage in the middle cerebral artery occlusion. Brain Research., 2000; 858(1): 55-60.
CrossRef - Korkmaz B, Horwitz M. S, Jenne D. E and Gauthier F. Neutrophil Elastase, Proteinase 3 and Cathepsin G as therapeutic targets in human diseases. Pharmacological Reviews., 2010; 62: 726–59.
CrossRef - Konrad C, Nabavi D. G, Junker R, Dziewas R, Henningsen H and Stogbauer F. Spontaneous internal carotid artery dissection and alpha-1-antitrypsin deficiency. Acta. Neurol. Scand., 2003; 107 (3):233-6.
CrossRef - Beheiri A, Langer C, During C, Krumpel A, Thedick S and Nowak-Gottl U. Role of elevated a2-macroglobulin revisited: results of a case-control study in children with symptomatic thromboembolism. Journal of Thrombosis and Haemostasis., 2007; 5: 1179–84.
CrossRef - Nezu T, Hosomi N, Aoki S, Deguchi K, Ichihara N, Ohyama H., et al. Alpha2-macroglobulin as a promising biomarker for cerebral small vessel disease in acute ischemic stroke patients. Journal of Neurology., 2013; 260(10): 2642-9.
CrossRef - Grau AJ, Seitz R, Immel A, Steichen-Wiehn C and Hacke W. Increased levels of leukocyte elastase in ischemic stroke and in subjects with vascular risk factors. Cerebrovasc Dis., 1995; 5:50-4.
CrossRef - Cojocaru I. M, Cojocaru M and Burcin C. Evaluation of granulocyte elastase as a sensitive diagnostic parameter of inflammation in first ischemic stroke. Rom. J. Intern. Med., 2006; 44: 317–21.
- Shi Z, Liu S, Guan Y, Zhang M, Lu H, Yue W., et al. Changes in total homocysteine levels after acute stroke and recurrence of stroke. Sci. Rep., 2018; 8(1):6993.
CrossRef - Joshi M. B, Baipadithaya G, Balakrishnan A, Hegde M, Vohra M, Ahamed R., et al. Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci. Rep., 2016; 6: 36362.
CrossRef - Baszczuk A and Kopczynski Z. Hyperhomocysteinemia in patients with cardiovascular disease. Postepy. Hig. Med. Dosw., 2014; 68:579.
CrossRef - Bieth J, Spiess B and Wermuth C. G. The synthesis and analytical use of a highly sensitive and convenient substrate of elastase. Biochem. Med., 1974; 11: 350-7.
CrossRef - Joshi M. B, Lad A, Prasad S. B, Balakrishnan A, Lingadakai R and Kapaettu S. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS letters. 2013; 587.14: 2241-6.
CrossRef - Man S, Ubogu E. E and Ransohoff R. M. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain. Pathol., 2007; 17:243–50.
CrossRef - Ishikawa N, Oda M, Kawaguchi M, Tsunezuka Y and Watanabe G. The effects of a specific neutrophil elastase inhibitor (ONO-5046) in pulmonary ischemia-reperfusion injury. Transpl. Int., 2003; 16:341–6.
CrossRef - Cojocaru I. M, Cojocaru M and Burcin C. Evaluation of granulocyte elastase as a sensitive diagnostic parameter of inflammation in first ischemic stroke. Rom. J. Intern. Med., 2006; 44: 317–21.
- Allen C. L and Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. stroke., 2009; 4(6):461-70.
CrossRef - Sayyed A. K, Despande K. H, Suryakar A. N, Ankush R. D and Katkam R. V. Oxidative Stress and Serum Α1 – Antitrypsin in Smokers. Indian Journal of Clinical Biochemistry., 2008; 23 (4):375-7.
CrossRef - Meschia J. F. Alpha-1 antitrypsin dysfunction and large artery stroke. Proc. Natl. Acad. Sci., 2017; 114(14):3555-7.
CrossRef - Malik R, Dau T, Gonik M, Shivakumar A, Deredge D. J, Edeleva E. V., et al. Common coding variant in SERPINA1 increases the risk for large artery stroke. PNAS., 2017; 114(14):3613-8.
CrossRef - Lehotsky J. J, Tothova B, Kovalska M, Dobrota D, Benova A, Kalenska D., et al. Role of Homocysteine in the Ischemic Stroke and Development of Ischemic Tolerance. Neurosci., 2016; 10: 538.
CrossRef - Tao Z, Yuan J, Shuhua Z, Tingting T, Yan C, Xiaoming S., et al. The association between homocysteine and ischemic stroke subtypes in Chinese. Medicine., 2020; 99: e19467.
CrossRef - Jourdheuil-Rahmani D, Rolland P. H, Rosset E, Branchereau A and Garcon D. Homocysteine induces synthesis of a serine elastase in arterial smooth muscle cells from multi-organ donors. Cardiovascular Research., 1997; 34: 597–02.
CrossRef

