
Nataliya Kitsera1,4 , Zoriana Osadchuk2, Mariya Dushar2, Oleh Hnateiko1,2, Nadiya Helner3, Maya Bondarenko4, Roman Bahrynovskyi4, Olha Dorosh5 and Ruslan Kozovyi4
, Zoriana Osadchuk2, Mariya Dushar2, Oleh Hnateiko1,2, Nadiya Helner3, Maya Bondarenko4, Roman Bahrynovskyi4, Olha Dorosh5 and Ruslan Kozovyi4
1Department of Birth Defects, Institute of Hereditary Pathology, National Academy of Medical Sciences of Ukraine, Lviv, Ukraine.
2Department of Clinical Genetics, Institute of Hereditary Pathology, National Academy of Medical Sciences of Ukraine, Lviv, Ukraine
3Medical Genetic Center, Institute of Hereditary Pathology, National Academy of Medical Sciences of Ukraine, Lviv, Ukraine
4Department of Medical Biology and Medical Genetics, Ivano-Frankivsk National Medical University, Ivano-Frankivsk, Ukraine.
5Department of Hematology and Іntensive Chemotherapy, Western Ukrainian Specialized Children's Medical Centre, Lviv, Ukraine
Corresponding Author e-mail: nkitsera@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/2567
Abstract
Rett syndrome is one of the most common causes of mental retardation in girls. The aim of our work was to study a spectrum of genetic heterogeneity and various clinical manifestations of Rett syndrome in girls Western Ukraine. Materials and methods: there were used clinical, molecular and genetic methods. We observed seven girls with Rett syndrome aged from 6 months to 15 years who were diagnosed and followed-up at the Institute of Hereditary Pathology, National Academy of Medical Sciences of Ukraine, Lviv for three years (2019–2021) and underwent molecular genetic analyses confirmed by next-generation sequencing. Results: In this study, patients with Rett syndrome had individual clinical heterogeneity and age variability due to different mutations. Mental retardation was not observed among siblings in families with Rett syndrome. We identified seven different pathogenic mutations among seven girls, including two deletions and one duplication of the MECP2 gene. Microcephaly was observed in two girls with MECP2 c.880C>T (p.Arg294*) and MECP2 Gain (Entire coding sequence) at birth. The following developmental disabilities were found in five girls: lack of independent sitting, lack of independent gait (regression of development). Among musculoskeletal disorders, there were diagnosed scoliosis, X-shaped deformation of the lower extremities and muscular hypotonia. A two-year-old girl with Rett syndrome, along with other clinical symptoms, had breathing problems - hyperventilation (rapid shallow breathing). Conclusions: in cases with unknown cause of delayed developmental disability and mental retardation the patients should be referred for medical genetic counselling.
Keywords
Clinical Manifestation; MECP2 Gene; Rett Syndrome; Ukraine
Download this article as:| Copy the following to cite this article: Kitsera N, Osadchuk Z, Dushar M, Hnateiko O, Helner N, Bondarenko M, Bahrynovskyi R, Dorosh O, Kozovyi R. Spectrum of Mutations and Clinical Manifestations of Rett Syndrome in Girls from Western Ukraine. Biomed Pharmacol J 2022;15(4). |
| Copy the following to cite this URL: Kitsera N, Osadchuk Z, Dushar M, Hnateiko O, Helner N, Bondarenko M, Bahrynovskyi R, Dorosh O, Kozovyi R. Spectrum of Mutations and Clinical Manifestations of Rett Syndrome in Girls from Western Ukraine.Biomed Pharmacol J 2022;15(4). Available from: https://bit.ly/3Hw5dfi |
Introduction
Rett syndrome is one of the most common causes of mental retardation in girls. About 6,500 girls with Rett syndrome are born worldwide each year. This syndrome affects all races and ethnic groups. Rett syndrome affects approximately 1:10,000-15,000 female births but differs in different countries and studies1-3.
Rett syndrome is an X-linked neurodevelopmental disorder with with associated multisystem comorbidities:progressive neurological symptoms, hypotonia, involuntary movement, a severe mental retardation, and epilepsy. The main clinical features are severe intellectual disability, loss of hand and speech skills, microcephaly, seizures, epilepsy, respiratory and motor abnormalities, but neurological symptoms appear first4,5.
Diagnosis depends on clinical presentation with or without a pathogenic mutation of the MECP2 or other genes. Genetic testing is commonly done as a part of the current diagnostic pathway, but many children may not have undergone genetic testing in Ukraine6. The clinical characteristics of Rett syndrome first appear in early childhood. That is the second leading genetic cause of intellectual disability in girls after Down syndrome. Most frequently caused by pathogenic variants in the MECP2 (methyl CpG binding protein 2) gene. More than 10,000 mutations in the MECP2 gene located on the X chromosome have been identified so far7,8.
In our paper, we investigated the variety of mutations and clinical symptoms in girls with Rett syndrome residing in Western Ukraine.
Materials and Methods
There were used clinical, molecular. Inclusion criteria were patients with gait abnormalities, partial or complete loss of acquired purposeful hand skills, spoken language, and stereotypic hand movements. Exclusion criteria were patients with abnormal psychomotor development within the first six months of life, brain injury secondary to trauma (peri‐ or postnatally). All the patients gave informed consent after the purpose and nature of the study were explained and their data were disclosed. All study personnel data were anonymized prior to the analysis.
This research was conducted in accordance with the provisions of the WMA Declaration of Helsinki. Clinical manifestations and molecular results were collected from the medical records of the patients. Informed written consents were taken from the parents of all participants. Ethical approval for this study was obtained by the Ethics Committee on May 03, 2022. Molecular genetic tests were done by next-generation sequencing (NGS, INVITAE Laboratory, USA). The study included girls with Rett syndrome with an identified MECP2 gene mutation.
Results
We observed seven girls with Rett syndrome aged from 6 months to 15 years from Western Ukraine who were diagnosed and followed-up at the Institute of Hereditary Pathology, National Academy of Medical Sciences of Ukraine, Lviv for three years (2019–2021) and underwent molecular genetic analyses confirmed by NGS.
Our patients had symptoms typical for Rett syndrome and the diagnosis was confirmed by molecular genetic analysis. No consanguineous marriages were recorded in the families of the surveyed probands. In families where the proband had siblings, no similar pathology was observed. In our group the clinical manifestations and pathogenic gene spectrum between female patients were different (table 1).
The onset of clinical manifestations was noted by parents at an early age – from 6 months to 2 years. Age at diagnosis of Rett syndrome was younger in girls with lack of eye contact, stereotypical movements, and lack of independent sitting. At the same time, the difference in the time of diagnosis of Rett syndrome in girls is quite large. A 2-year-old girl (case 6) who was diagnosed with c.502C>T (p.Arg168*) mutation of the MECP2 was the youngest in our group. Only in one case, in adolescence, a 15-year-old girl (case 1) was diagnosed with Rett syndrome using molecular genetic analysis – MECP2 Gain (Entire coding sequence).
In our study, Rett syndrome had a complex and multifaceted clinical appearance. The girls with the indicated neurological symptoms were observed by a neurologist for a long time, but the treatment had no effect, and the parents began to observe a regression of acquired skills. Among the neurological manifestations of Rett syndrome, . Microcephaly was observed in two girls (cases 1, 4) with Rett syndrome with MECP2 c.880C>T (p.Arg294*) and MECP2 Gain (Entire coding sequence) at birth.
The following developmental disabilities were found in five girls: lack of independent sitting, lack of independent gait (regression of development). Among musculoskeletal disorders, there were diagnosed scoliosis, X-shaped deformation of the lower extremities and muscular hypotonia.
Our youngest female (case 2) aged 6 months had the MECP2 c.473C>T (p.Thr158Met) variant with clinical symptoms of lack of eye contact, stereotypic movements, lack of independent sitting, muscular hypotension which occurred after a short period of initially normal development. Among other symptoms, a 2-year-old girl (case 6) girl with MECP2 c.502C>T (p.Arg168*) had breathing problems: hyperventilation (rapid shallow breathing).
Most of our girls had verified neurological symptoms and the diagnosis of Rett syndrome was finally made only after consultation with a geneticist and molecular genetic confirmation.
Molecular genetic analysis was performed to all patients diagnosed with Rett syndrome in our series (Table 1): in cases 4 and 6, the girls had c.880C>T (p.Arg294*) and c.502C>T (p.Arg168*) mutations, respectively. These mutations are frequently seen in typical Rett syndrome. Duplication of all gene were identified in case1 – MECP2 Gain (Entire coding sequence). In cases 7 and 5, – c.1157_1197 del (p.Leu386 Hisfs*5) and c.696del (p.Lys233Argfs*15).
Among other concomitant manifestations, in one girl aged 6 years (case 7), diffuse goiter, adiposity, rhinitis, and otitis were observed from an early age.
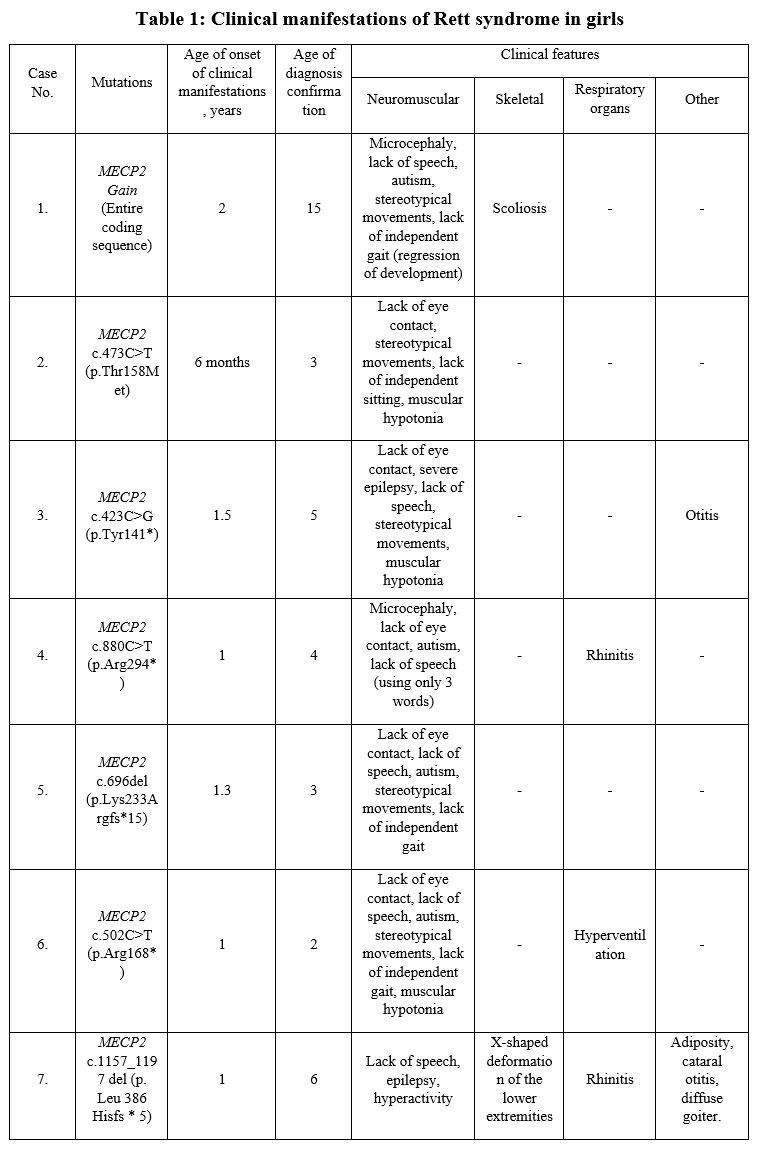 |
Table 1: Clinical manifestations of Rett syndrome in girls |
Discussion
In this study, we reported the results of the genetic analysis of the MECP2 gene and clinical features in girls with classical Rett syndrome. Rett syndrome caused by mutations in the MECP2 gene is one of the most frequent reasons for mental retardation in females. A genetic confirmation typically follows the clinical diagnosis of Rett syndrome.
Mutations of the gene encoding methyl‐CpG‐binding protein 2 (MeCP2), located at chromosome Xq28, most often cause Rett syndrome. Therefore, this defect disrupts normal nerve cell function and causes mental retardation.
In our study, MECP2 mutations are connected with a wide spectrum of phenotypes, including classical Rett syndrome such as an early‐onset seizure type to preserved speech variant, and other kinds of intellectual disabilities, such as autism. According to researchers, epilepsy of varying degrees is often diagnosed in patients with Rett syndrome9. In our study, two girls had epilepsy of varying degrees and the selected treatment lasted for years. Moreover, a 5-year-old girl with a mutation in MECP2 c.423C> G (p.Tyr141*) had severe epilepsy.
According to many researchers, the most common pathogenic variants of the MECP2 gene are p.Arg168*, p.Arg294*, p.Arg255*, p.Arg270*, p.Arg106*, p.Arg133*, p.Arg158* and p.Arg306*10,11..These variants are responsible for nearly 50% of the patients with Rett syndrome10. In our patients, there were different pathogenic variants of the MECP2 gene, including p.Arg168* and p.Arg294*.
Deletions involving one or more exons of the MECP2 gene were reported in more than 10% of patients with suspected Rett syndrome from Turkey, but in our study, we diagnosed them in two cases (28.6%)12.
An observation of seven girls with Rett syndrome, mental and developmental regression, stereotypic hand movements suggested that female Rett syndrome may survive. In our study, in one girl, the diagnosis of Rett syndrome was confirmed by molecular genetic research at the age of 15 years.
Many physicians report respiratory disorders in children with Rett syndrome13,14. In our group,
Although most research is focused on the role of MECP2 in the central nervous system, the different clinical aspects in Rett syndrome patients underline the importance of MECP2 in another systems.
In summary, this study expands our knowledge of cases with the MECP2 gene mutations which may produce a spectrum of various clinical manifestations in females.
Conclusions
In this study, patients with Rett syndrome had individual clinical heterogeneity and age variability due to different mutations. In families where the proband had siblings, no similar pathology was observed.
We identified seven different pathogenic mutations among seven girls, including two deletions and one duplication of the MECP2 gene.
Microcephaly was observed in two girls with Rett syndrome with MECP2 c.880C>T (p.Arg294*) and MECP2 Gain (Entire coding sequence) at birth.
All female children with low intelligence, defined as having mental retardation, epilepsy, and autistic symptoms, should be suspected of having Rett syndrome.
In cases with unknown cause of delayed developmental disability and mental retardation, the patients should be referred for medical genetic counselling..
Authors’ Contributions
All authors participated in the research design, data analysis, and the writing of the manuscript. All authors approved the final version of the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Funding Sources
Authors state no funding involved.
References
- Meireles, P. T., Soares, M. B., Lacerda, D. R. N. B., Gomes, B. B. M., Carvalho, E. E. V. de Salge, A. K. M., Abdalla, G. K and Abdalla, D. R. Rett Syndrome in a Peruvian patient: A case report / Síndrome de Rett em um paciente peruano: Um relato de caso. Braz. J. Dev., 2020; 6(8): 63967–63974. https://doi.org/10.34117/bjdv6n8-722
CrossRef - Nasiri, J., Salehi, M., Hosseinzadeh, M., Zamani, M., Fattahpour, S., Aryani, O., Najafabadi, E. F., Jabarzadeh, M., Asadi, S., Gholamrezapour, T., Sedghi, M and Ghorbani, F. Genetic Analysis of MECP2Gene in Iranian Patients with Rett Syndrome. Iran. J. Child. Neurol., 2019; 13(3): 25-34.
- Zahorakova, D., Lelkova, P., Gregor,, Magner, M., Zeman, J and Martasek, P. MECP2mutations in Czech patients with Rett syndrome and Rett-like phenotypes: novel mutations, genotype – phenotype correlations and validation of high-resolution melting analysis for mutation scanning. J. Hum. Genet., 2016; 61: 617-625. https://doi.org/10.1038/jhg.2016.19
CrossRef - Anderson, A., Wong, K., Jacoby, P., Downs, J and Leonard, H. Twenty years of surveillance in Rett syndrome: what does this tell us? Orphanet. J. Rare. Dis., 2014; 9: 87. https://doi.org/10.1186/1750-1172-9-87
CrossRef - Xiol, C., Heredia, M., Pascual-Alonso, A., Oyarzabal, A and Armstrong, J. Technological Improvements in the Genetic Diagnosis of Rett Syndrome Spectrum Disorders. Int. J. Mol. Sci., 2021; 22(19): https://doi.org/10.3390/ijms221910375
CrossRef - Bzduch, V., Zahorakova, D., Grechanina, E., Zdibskaja, P., Goldfarb, I. G., Zeman, J and Martasek, P. A case of Rett syndrome from Ukraine – clinical diagnosis confirmed by mutation analysis of the MECP2 gene. Bratisl. Lek. Listy., 2004; 105(9): 299-302.
- Ehrhart, F., Jacobsen, A., Rigau, M., Bosio, M., Kaliyaperumal, R., Laros, J. F. J., Willighagen, E. L., Valencia, , Roos, M., Capella-Gutierrez, S., Curfs, L. M. G and Evelo, C. T. A catalogue of 863 Rett-syndrome-causing MECP2mutations and lessons learned from data integration. Sci. Data., 2021; 8: 10. https://doi.org/10.1038/s41597-020-00794-7
CrossRef - Gold, W. A., Krishnarajy, R., Ellaway, C and Christodoulou, J. Rett Syndrome: A Genetic Update and Clinical Review Focusing on Comorbidities. ACS. Chem. Neurosci., 2018; 9(2): 167-176. https://doi.org/10.1021/acschemneuro.7b00346
CrossRef - Operto, F. F., Mazza, R., Pastorino, G. M. G., Verrotti, A and Coppola, Epilepsy and genetic in Rett syndrome: A review. Brain. Behav., 2019; 9(5): e01250. https://doi.org/10.1002/brb3.1250
CrossRef - Downs, J., Stahlhut, M., Wong, K., Syhler, B., Bisgaard, A. M., Jacoby, P., & Leonard, H. (2016). Validating the Rett Syndrome Gross Motor Scale. PloS one, 11(1), e0147555. https://doi.org/10.1371/journal.pone.0147555
CrossRef - Fang, X., Butler, K. M., Abidi, F., Gass, J., Beisang, A., Feyma, T., Ryther, R. C., Standridge, S., Heydemann, P., Jones, M., Haas, R., Lieberman, D. N., Marsh, E. D., Benke, T. A., Skinner, S., Neul, J. L., Percy, A. K., Friez, M. J., & Caylor, R. C. (2022). Analysis of X-inactivation status in a Rett syndrome natural history study cohort. Molecular genetics & genomic medicine, 10(5), e1917. https://doi.org/10.1002/mgg3.1917
CrossRef - Hazan, Z., Gorsoy, S., Unalp, A and Xilmax, U. Clinical evolution of patients with classical Rett syndrome and MECP2 gene analyse. DEU. Tip. Derq., 2021; 35(1): 87-97. https://doi.org/10.5505/deutfd.2021.50133
CrossRef - Sarber, K. M., Howard, J. J. M., Dye, T. J., Pascoe, J. E and Simakajornboon, Sleep-disordered breathing in pediatric patients with Rett syndrome. J. Clin. Sleep. Med.,2019; 15(10): 1451-1457. https://doi.org/10.5664/jcsm.7974
CrossRef - Ramirez, J. M., Karlen-Amarante, M., Wang, J. J., Bush, N. E., Carroll, M. S., Weese-Mayer, D. E., & Huff, A. (2020). The Pathophysiology of Rett Syndrome With a Focus on Breathing Dysfunctions. Physiology (Bethesda, Md.), 35(6), 375–390. https://doi.org/10.1152/physiol.00008.2020
CrossRef

