
Manuscript accepted on :05-11-2021
Published online on: 14-12-2021
Plagiarism Check: Yes
Reviewed by: Dr. Salman Ahmed
Second Review by: Dr. Sohayla Mohamed Elsherbini Attalla
Final Approval by: Dr. Ian James Martin
Malathi Kullappan1 , Jenifer M Ambrose1 and Surapaneni Krishna Mohan2*
, Jenifer M Ambrose1 and Surapaneni Krishna Mohan2*
1Department of Research, Panimalar Medical College Hospital and Research Institute, Varadharajapuram, Poonamallee, Chennai - 600 123, India.
2Departments of Biochemistry, Clinical Skills and Simulation, Research, Panimalar Medical College Hospital and Research Institute, Varadharajapuram, Poonamallee, Chennai - 600 123, India.
Corresponding Author E-mail: krishnamohan.surapaneni@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/2291
Abstract
COVID-19, a pandemic disease caused by single-stranded RNA virus Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2). The structural spike (S) protein of SARS-CoV-2 plays a vital role in host cell entry, where the Angiotensin-Converting Enzyme-2 (ACE2) receptor of the human cell binds to the Receptor Binding Domain (RBD) region of the S1 domain and makes cell entry. The binding affinity of SARS-CoV-2-ACE2 is tenfold higher than the SARS-CoV-1-ACE2. Recent studies expose that the SARS-CoV-2 S D614G variant is highly infectious than D614 protein, also the D614G variant is highly stable than D614. So far, there is no effective viral-specific regimen for COVID-19. To overcome such problems, in our study, we have utilized the ZINC database to screen potent leads against the highly transmitting SARS-CoV-2 spike D614G protein, through a virtual screening procedure. We have applied three computational tools iGEMDOCK server, AutoDock version 4.2.6 and admetSAR to get active leads. The ZINC000150588351 (Elbasvir), ZINC000064540179 (Sofosbuvir analogue) and ZINC000137700912 (Sofosbuvir analogue) molecules have a greater binding affinity with the high binding energies of -8.22 kcal/mol, -8.13 kcal/mol and -7.64 kcal/mol respectively. The molecules ZINC000064540179 and ZINC000137700912 have high binding energy than their core molecule Sofosbuvir (ZINC100074252) of -4.06 kcal/mol. The ADMET prediction of these molecules reveals satisfactory human intestinal absorption and non-mutagenic property. Our results deliver valuable contributions to the design of inhibitors against COVID-19.
Keywords
Active lead; Binding Energy; COVID-19; Molecular docking; SARS-CoV-2; Virtual Screening
Download this article as:| Copy the following to cite this article: Kullappan M, Ambrose J. M, Mohan S. K. Lead Identification for Severe Acute Respiratory Syndrome Coronavirus-2 Spike D614G Variant of COVID-19: A virtual Screening Process.Biomed Pharmacol J 2021;14(4). |
| Copy the following to cite this URL: Kullappan M, Ambrose J. M, Mohan S. K. Lead Identification for Severe Acute Respiratory Syndrome Coronavirus-2 Spike D614G Variant of COVID-19: A virtual Screening Process. Biomed Pharmacol J 2021;14(4).Available from: https://bit.ly/3dQLmYk |
Introduction
COVID-19, a pandemic disease which was found in December 2019 in Wuhan, China was caused by Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2).1,2 The World Health Organization (WHO) has named this novel CoV as “2019-nCoV” in January 2020. Later, it was found that the 2019-nCoV has a 78% sequence identity with SARS-CoV-1. Hence, the International Virus Classification Commission categorized 2019-nCoV as SARS-CoV-2 in February 2020.2 CoVs fit under Coronaviridae family and Nidovirales order. SARS-CoV-2 grouped under beta CoV genera,3 it is more infectious and has an increased mortality rate.4 SARS-CoV-2 is a single-stranded RNA virus, which encodes structural (Spike (S), Envelope (E), Membrane (M) and Nucleocapsid (N)) and non-structural (replicase polyproteins) proteins.5 Here, the structural proteins S, E and M are bound to the membrane, while the N protein is situated inside the virions in combination with RNA6. The glycoprotein S is responsible for the recognition, attachment and host cell entry of SARS-CoV-2. Also, it act as an antigenic determinant and plays a major role in antibody neutralization.7 Hence, it is considered as the main target in CoV therapeutic research.8,9 In humans, CoV infection is initiated by the interaction of S protein of SARS-CoV-2 with the Angiotensin-Converting Enzyme-2 (ACE-2) (play an essential role in angiotensin maturation), a metalloprotease enzyme found in human lung cells and then begin to release the viral RNA into the human cell. During the host cell entry, trimeric S protein is sliced into S1 and S2 domains.2 This S1 domain has the receptor-binding domain (RBD), through which the SARS-CoV-2 is connected with the ACE-2 receptor.1 The S2 domain initiates the fusion of viral membranes.10 Few other human proteases namely, plasmin, trypsin, HAT, cathepsin L, TMPRSS2, TMPRSS4 and TMPRSS11a encourage S protein cleavage to promote attachment and fusion.8,11 Recent studies reveal that SARS-CoV-2 isolates have mutations in many genomic parts. Such one is found in the S1 domain of the S protein of SARS-CoV-2. In the S1 domain, at 614 positions the amino acid Aspartic acid (D) is replaced with Glycine (G).12 The literature evidence shows that the transmission of SARS-CoV-2 S proteins with D614G mutation is highly efficient than the D614 S protein. Such higher infectivity of D614G S protein is further confirmed by the in-vitro studies carried out with the epithelial cells of lung and colon, reveals the 4 fold increased infectivity of D614G S protein.13,14 The D614G S protein has more stability than the D614 S protein.12 Currently, many Food and Drug Administration (FDA) approved antiviral drugs have been proposed to treat COVID-19, remdesivir, remdesivir/chloroquine, lopinavir/ritonavir, ribavirin/interferons.5,15,16 These drugs are not viral-specific therapeutic medications for SARS-CoV-2. Such situations urge us to design a potential drug against SARS-CoV-2. Worldwide many clinical trials are in progress to identify potential drugs for COVID-19, aiming at different antiviral protein targets. In our study, we have screened currently existing antiviral drugs and their analogues (total of 500 compounds) against the highly transmitting SARS-CoV-2 S D614G protein, through virtual screening and molecular docking procedure. The resultant lead compounds were subjected to Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) property prediction to understand the oral bioavailability of the compounds. Our results will be useful for the researchers in the COVID-19 drug screening process.
Materials and Methods
Protein Preparation and active site prediction
The protein structure of the SARS-CoV-2 S D614G variant was recovered from the protein data bank (PDB),17 PDB ID: 6XS6.14 It is a homo-trimer, which has 3 identical chains, for our virtual screening and molecular docking purpose only A chain was utilized. Since the SARS-CoV-2 S has S1 and S2 domain, the D614G mutant was present at the C-terminal end. Hence, the S1 domain alone was selected for the current study. The missing RBD region and other residues of the structure were constructed using MODELLER 9.1618 and evaluated by the PROCHECK19 and ProSA server20. Finally, the modelled structure was energy minimized by the PDB viewer and used for virtual screening. The active site residues for virtual screening and docking studies were collected from the literature.
Protein stability prediction
The stability of the SARS-CoV-2 S D614G variant was analyzed by the DUET server.21 It is a support vector machine (SVM), which calculates the free energy changes (ΔΔG values) on missense mutation with a secondary structure as well as a pharmacophore vector. Such prediction is carried out through SDM (it utilizes environment-specific substitution tables (ESSTs) with constrained conformation) and mCSM (using graph-based structural signatures) methods.22
Ligands
Currently existing antiviral drugs namely, remdesivir, lopinavir, darunavir, amprenavir, rupintrivir, sofosbuvir, adefovirdipivoxil, famciclovir, tecovirimat, darunavir, zidovudine, dolutegravir, entecavir, bictegravir, oseltamivir, emtricitabine, zalcitabine, didanosine, elbasvir, simeprevir, ritonavir, tipranavir, nelfinavir, dolutegravir, tenofovir-disoproxil, baloxavir-marboxil, letermovir, maraviroc and their analogues, a total of 500 compounds were collected from the ZINC database (Table 1).23
Table 1: Currently existing antiviral drugs
| S.No: | Antiviral drugs | Description |
| 1. | Remdesivir | active against MERS-CoV, Ebola virus and SARS-CoV |
| 2. | Lopinavir | lopinavir/ritonavir used against human immunodeficiency virus (HIV) infection |
| 3. | Darunavir | HIV-1 protease inhibitor |
| 4. | Amprenavir | protease inhibitor used against HIV infection |
| 5. | Rupintrivir | inhibitor against human rhinoviruses |
| 6. | Sofosbuvir | inhibitor of the hepatitis C virus (HCV) RNA polymerase |
| 7. | Adefovirdipivoxil | reverse transcriptase inhibitor, have activity against HBV |
| 8. | Famciclovir | Active against herpes simplex 1 and 2 |
| 9. | Tecovirimat | Used against orthopoxviruses |
| 10. | Darunavir | HIV-1 protease inhibitor |
| 11. | Zidovudine | inhibits DNA polymerase and leads to cell death |
| 12. | Dolutegravir | used against HIV-1infections |
| 13. | Entecavir | activity against HBV |
| 14. | Bictegravir | integrase strand transfer inhibitor, HIV |
| 15. | Oseltamivir | have antiviral activity |
| 16. | Emtricitabine | Inhibits HIV reverse transcriptase |
| 17. | Zalcitabine | inhibits γ-DNA |
| 18. | Didanosine | Inhibits HIV reverse transcriptase |
| 19. | Elbasvir | Used against HCV genotype 1 and 4 |
| 20. | Simeprevir | HCV protease inhibitor |
| 21. | Ritonavir | Inhibitor of antiretroviral protease, HIV-1 |
| 22. | Tipranavir | Inhibitor of antiretroviral protease, Used against HIV |
| 23. | Nelfinavir | Used against HIV infection |
| 24. | Nelfinavir | Used against HIV infection |
| 25. | Dolutegravir | Used against HIV-I infection |
| 26. | Tenofovir-disoproxil | Used to treat HIV and HBV |
| 27. | Baloxavir-marboxil | Used against influenza A and B infections |
| 28. | Letermovir | Activity against cytomegalovirus |
| 29. | Maraviroc | Used against HIV |
Virtual screening
In the drug discovery process, virtual screening has become a potent in-silico method to screen millions of small molecules for the lead with preferred properties. The main purpose of the virtual screening process is to increase drug discovery and not to substitute in-vitro or in-vivo analysis. In our current study, we have implemented structure-based virtual screening and employed ZINC database for small molecules, iGEMDOCK server (Hsu et al., 2011) for virtual screening, AutoDock version 4.2.619 for molecular docking and admetSAR for ADMET property prediction (Figure 1).24
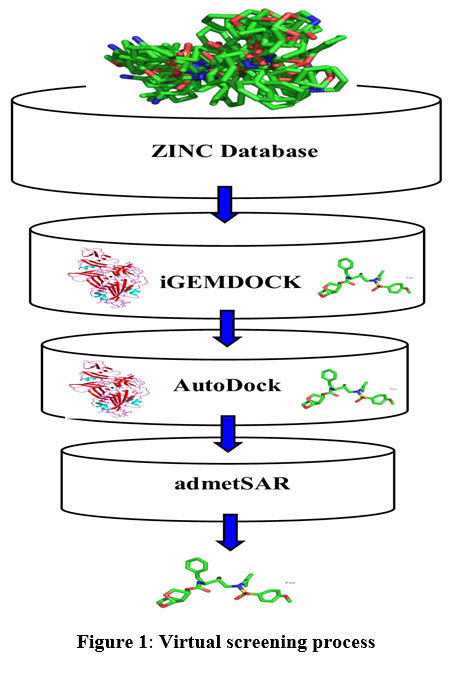 |
Figure 1: Virtual screening process |
iGEMDOCK server
It is a user-friendly graphical environment, which provides space for ligand and target protein preparation to docked pose analysis. The pharmacological interactions reveal the protein-ligand binding mechanism. The iGEMDOCK is useful for both post-screening analysis and pharmacological interactions. The target protein and ligands were submitted in PDB format. The iGEMDOCK employs an empirical scoring system and measures the total energy between the protein and ligand through van der Waals (vdW), H-bond and electrostatic (Elec) interactions (Fitness = vdW + H-bond + Elec, where, Fitness-total energy).25
AutoDock version 4.2.6
AutoDock software (version 4.2.6) was used for the current study. It functions based on the Lamarckian genetic algorithm (LGA), a fusion of Genetic Algorithm (GA), hybrid GA (local search hybridized with GA) and Monte Carlo Simulation. Through the graphical user interface of the AutoDock tool, the polar hydrogen (H) atoms were added to the SARS-CoV-2 S D614G structure (PDB format) and the non-polar H atoms were merged. The atoms were assigned with the Kollman partial charges. Then, the refined protein structure was saved in PDBQT format. Similarly, the ligand molecules were set with the torsion angles for flexible docking, later the ligand was saved in PDBQT format. The docking procedure was based on the specific binding site regions, hence the grid box was set to minimize the run time. LGA is the most widely used method for docking studies. The final docked conformations were analyzed through the Auto-dock tool.19
ADMET property prediction
In the process of drug discovery and development, ADMET prediction plays an essential part. The drug discovery process includes particular disease and drug target, lead compound optimization, preclinical studies (in-vivo and in-vitro), filing for Food and Drug Administration (FDA), clinical studies Phase I, phase II and Phase III, New Drug Application (NDA) applied for FDA approval, a drug approved for marketing. The drug discovery process is highly a time-consuming one. Although, lots of active leads have been discovered, in recent years there is only less number of drugs have been got approved. ADMET property deficiencies of those drugs might be the reason for such stagnation. To overcome such problems, the ADMET properties of the lead compounds resulted from the virtual screening process were projected through the online server admetSAR, which predicts the drug-likeness, pharmacokinetic and pharmacodynamic properties.24
Results and Discussion
Protein structure validation
MODELLER has created five protein models with different Discrete Optimized Protein Energy (DOPE). The model with a decreased DOPE score was selected as the best model (Figure 2). The PROCHECK server has screened the stereochemical property of the constructed model with Ramachandran Plot. It indicates that amino acids are located in the favored regions. PROSA shows the Z-score value of -8.3. Both PROCHECK and ProSA further confirmed the reliability of the constructed model. The active site residues of the SARS-CoV-2 S protein which are playing an essential role in protein-ligand binding namely, GLN493, LEU452, LEU455, LEU492, LYS458, PHE486, PHE490, SER494 and TYR489 are collected from the literature.
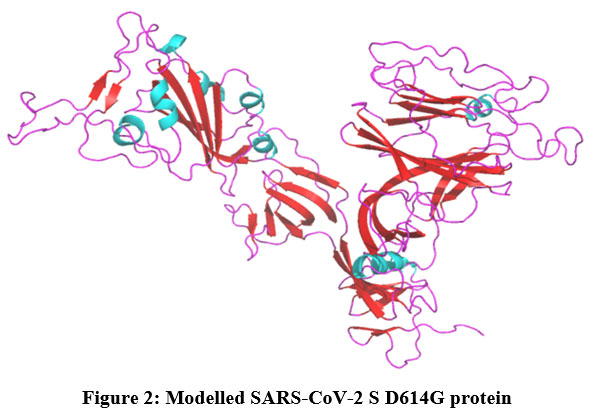 |
Figure 2: Modelled SARS-CoV-2 S D614G protein. |
Protein stability prediction
The DUET server has predicted the stability of the SARS-CoV-2 S D614G mutation by the free energy changes. DUET has shown the ΔΔG value of 0.475 kcal/mol (Stabilizing) (Table 2). The complementary methods of the DUET, the SDM and mCSM have the ΔΔG values of 2.33 kcal/mol (Stabilizing) and -0.21 kcal/mol (Destabilizing). The ΔΔG values of DUET and SDM reveal the stability of the SARS-CoV-2 S D614G mutation. Our results resemble the in-vitro studies carried out by Zhang et al., 2020.12
Table 2: DUET stability prediction of SARS-CoV-2 S D614G mutation
| S.no: | Name of the protein | Mutation
position |
Amino acid change | DUET
(ΔΔG) (kcal/mol) |
SDM
(ΔΔG) (kcal/mol) |
mCSM
(ΔΔG) (kcal/mol) |
| 1. | SARS-CoV-2 S | 614 | ASP to GLY | 0.475
(Stabilizing) |
2.33
(Stabilizing) |
-0.21
(Destabilizing) |
ΔΔG – free energy
Virtual screening
To identify the active lead against SARS-CoV-2 S D614G, a total of 500 small molecules were submitted to iGEMDOCK, this has provided the total energy (vdW + H-bond + Elec) of SARS-CoV-2 S D614G – ZINC compounds in kcal/mol. From the screened 500 compounds, the best 50 compounds with high total energy scores were selected and it is tabulated in Table 3. This reveals that the ZINC compounds ZINC000150588351 to ZINC000101802366, have the total energies in the range of -119.61 to -103.1 kcal/mol, ZINC001772647545 to ZINC001560410199 compounds with -98.67 to -90.4 kcal/mol range, compounds ZINC001220115960 to ZINC000100003902 within the range of -89.15 to -80.25 kcal/mol and compounds ZINC000100368814 to ZINC000096169912 scores ranges from -79.50 to -70.22 kcal/mol respectively. From these 50 compounds, the compounds ZINC000150588351, ZINC000064540179, ZINC000166442088, ZINC000137700912, ZINC001772647544 and ZINC000101802366 are considered to be the best binding energy compounds. These are subjected to docking procedures with AutoDock version 4.2.6. After molecular docking with AutoDock, the compounds ZINC000150588351 (Elbasvir), ZINC000064540179 and ZINC000137700912 (Sofosbuvir analogues) are selected as the best binding energy compounds (Table 4). They have the binding energies of -8.22 kcal/mol, -8.13 kcal/mol and -7.64 kcal/mol respectively. The amino acid residues ALA352 (2 bonds), ARG466, ASN354 and LEU492 of SARS-CoV-2 S D614G are involved in the H-bond interaction with ZINC000150588351.
Table 3: iGEMDOCK scores for virtual screening of SARS-CoV-2 S D614G and ZINC compounds (kcal/mol)
| S. No. | Compound ID | vdW | H-Bond | Elec | Total Energy |
| 1 | ZINC000150588351 | -90.3 | -29.32 | 0 | -119.61 |
| 2 | ZINC000064540179 | -90.17 | -29.43 | 0 | -119.6 |
| 3 | ZINC000137700912 | -87.2 | -18.75 | 0 | -105.95 |
| 4 | ZINC001772647544 | -89.1 | -13.76 | 0 | -102.86 |
| 5 | ZINC000166442088 | -92.12 | -16.23 | 0 | -108.35 |
| 6 | ZINC000101802366 | -88.99 | -14.11 | 0 | -103.1 |
| 7 | ZINC001772647545 | -81.99 | -16.68 | 0 | -98.67 |
| 8 | ZINC000028232111 | -79.97 | -18.64 | 0 | -98.61 |
| 9 | ZINC001772610688 | -70.04 | -26.1 | 0 | -96.14 |
| 10 | ZINC001560410107 | -79.4 | -16.26 | 0 | -95.66 |
| 11 | ZINC000097943124 | -91.49 | -2.29 | 0 | -93.78 |
| 12 | ZINC000003802690 | -62.28 | -30.94 | 0 | -93.22 |
| 13 | ZINC001220116037 | -75.95 | -17.93 | 0 | -93.88 |
| 14 | ZINC000195806232 | -83.74 | -8.67 | 0 | -92.41 |
| 15 | ZINC000100481369 | -72.82 | -18.41 | 0 | 91.23 |
| 16 | ZINC000064526913 | -74.02 | -17.11 | 0 | -91.13 |
| 17 | ZINC000003929038 | -71.84 | -19.41 | 0 | -91.25 |
| 18 | ZINC000166442021 | -65.48 | -26.39 | 0 | -91.88 |
| 19 | ZINC001220115824 | -75.41 | -15.56 | 0 | -90.96 |
| 20 | ZINC000097943123 | -81.38 | -9.0 | 0 | -90.48 |
| 21 | ZINC001560410199 | -87.28 | -3.11 | 0 | -90.4 |
| 22 | ZINC001220115960 | -79.71 | -9.45 | 0 | -89.15 |
| 23 | ZINC000003930376 | -67.72 | -21.09 | 0 | -88.81 |
| 24 | ZINC000003955219 | -80.99 | -7.64 | 0 | -88.63 |
| 25 | ZINC000095559650 | -61.68 | -25.87 | 0 | -87.55 |
| 26 | ZINC000014947885 | -56.1 | -30.98 | 0 | -87.08 |
| 27 | ZINC000100074252 | -70.51 | -16.43 | 0 | -86.94 |
| 28 | ZINC000001530635 | -68.07 | -18.7 | 0 | -86.77 |
| 29 | ZINC000013597823 | -67.93 | -12.86 | 0 | -85.48 |
| 30 | ZINC000058581064 | -73.52 | -11.95 | 0 | -85.48 |
| 31 | ZINC000011682512 | -65.95 | -18.31 | 0 | -84.26 |
| 32 | ZINC000003915160 | -59.86 | -23.69 | 0 | -83.55 |
| 33 | ZINC000085548369 | -68.83 | -14.46 | 0 | -83.28 |
| 34 | ZINC000003779042 | -49.49 | -33.74 | -0.02 | -83.25 |
| 35 | ZINC000028525892 | -74.91 | -8.2 | 0 | -83.12 |
| 36 | ZINC000169289453 | -72.27 | -10.4 | 0 | -82.67 |
| 37 | ZINC001220115510 | -64.86 | -17.72 | 0 | -82.57 |
| 38 | ZINC001220115959 | -69.06 | -13.22 | 0 | -82.28 |
| 39 | ZINC001220116162 | -72.95 | -8.9 | 0 | -81.85 |
| 40 | ZINC000003629271 | -65.72 | -15.95 | 0 | -81.67 |
| 41 | ZINC000059402108 | -66.6 | -15.01 | 0 | -81.61 |
| 42 | ZINC001220115377 | -73.95 | -7.17 | 0 | -81.12 |
| 43 | ZINC000095936387 | -63.86 | -17.04 | 0 | -80.9 |
| 44 | ZINC000100003902 | -72.42 | -7.83 | 0 | -80.25 |
| 45 | ZINC000100368814 | -60.08 | -19.42 | 0 | -79.50 |
| 46 | ZINC000000039906 | -52.03 | -27.4 | 0 | -79.43 |
| 47 | ZINC000003929508 | -69.1 | -9.49 | 0 | -78.59 |
| 48 | ZINC001560410106 | -59.12 | -18.08 | 0 | -77.2 |
| 49 | ZINC000095917469 | -57.11 | -15.03 | 0 | -72.14 |
| 50 | ZINC000096169912 | -56.45 | -13.77 | 0 | -70.22 |
vdW- van der Waals; H-bond – Hydrogen bond; Elec – Electrostatic
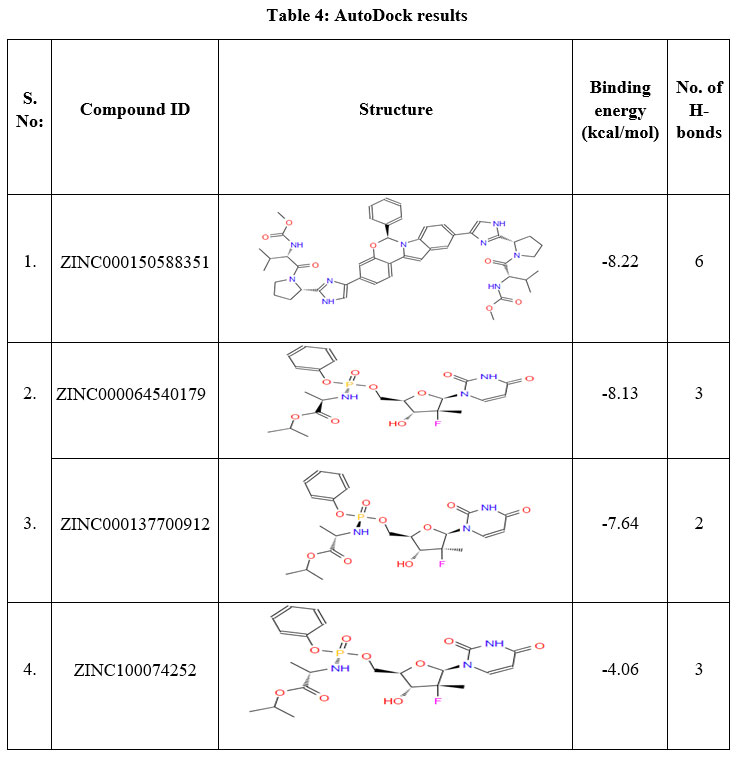 |
Table 4: AutoDock results |
A total of six H-bonds were found with bond distances of 3.4Å, 3.2Å, 2.5Å, 2.5Å and 2.4Å respectively (Figure 3 (A) & (B)). Compound ZINC000064540179 has three H-bonds with the residues ASN501, GLY496 and GLY502 of bond distances 2.2Å, 3.2Å and 2.2Å respectively (Figure 4 (A) & (B)). Similarly, compound ZINC000137700912 has formed two H-bonds with residues PHE490 (3.1Å) and SER494 (2.3Å) (Figure 5 (A) & (B)). The molecules ZINC000064540179 and ZINC000137700912 have high binding energy than the core molecule Sofosbuvir (ZINC100074252), which has a binding energy of -4.06 kcal/mol and 3 H-bond formations. The elbasvir, an antiviral drug that inhibits NS5A and prevents the replication of RNA and assembly of virions in hepatitis C virus (HCV).
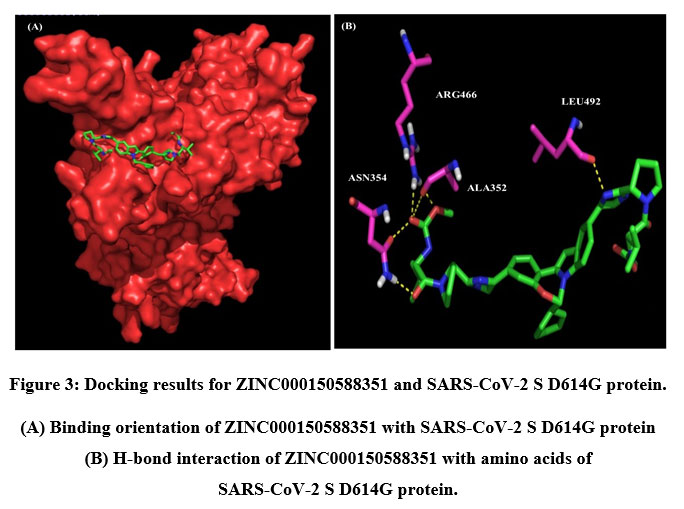 |
Figure 3: Docking results for ZINC000150588351 and SARS-CoV-2 S D614G protein. |
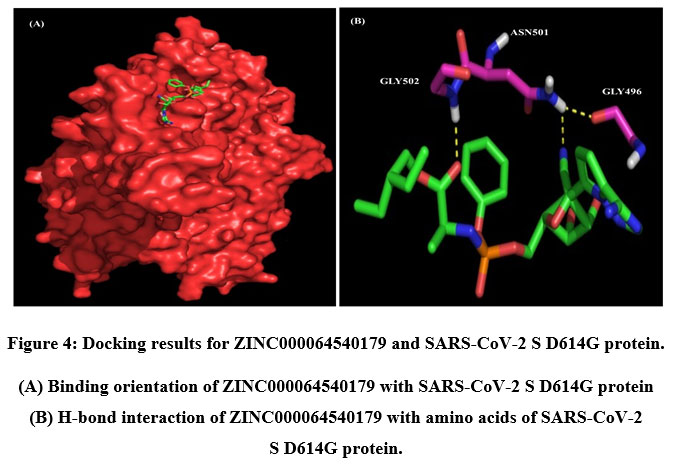 |
Figure 4: Docking results for ZINC000064540179 and SARS-CoV-2 S D614G protein. |
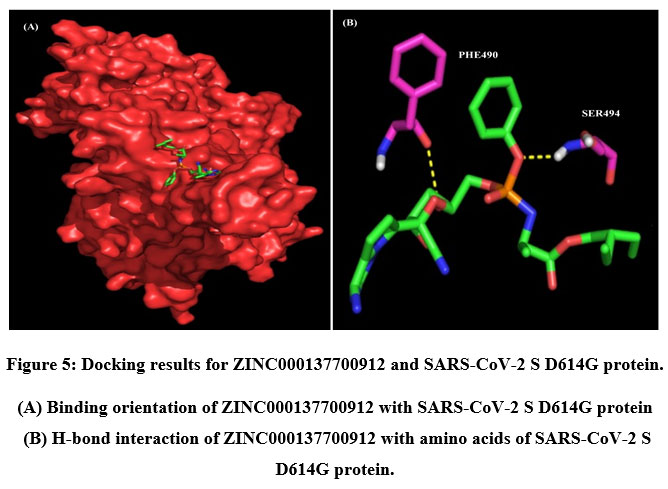 |
Figure 5: Docking results for ZINC000137700912 and SARS-CoV-2 S D614G protein. |
The elbasvir/grazoprevir combination is used in the treatment of HCV genotypes 1 and 4.26 Similarly, sofosbuvir is also inhibiting NS5B polymerase in HCV.27 The compounds ZINC000150588351, ZINC000064540179 and ZINC000137700912 have high binding energy and high binding affinity with SARS-CoV-2 S D614G protein. ADMET property prediction results are shown in Table 5.
Table 5: ADMET prediction
| S. no. | Models | ZINC000150588351 | ZINC000064540179 | ZINC000137700912 |
| Absorption | ||||
| 1. | Blood-Brain-Barrier | BBB- | BBB- | BBB- |
| 2. | Human Intestinal Absorption | HIA+ | HIA+ | HIA+ |
| 3. | Caco-2 Permeability | Caco2- | Caco2- | Caco2- |
| 4. | P-glycoprotein substrate | Substrate | Non-substrate | Non-substrate |
| 5. | P-glycoprotein inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor |
| 6. | Renal Organic Cation Transporter | Non-inhibitor | Non-inhibitor | Non-inhibitor |
| Metabolism | ||||
| 7. | CYP450 2C9 Substrate | Non-substrate | Non-substrate | Non-substrate |
| 8. | CYP450 2D6 Substrate | Non-substrate | Non-substrate | Non-substrate |
| 9. | CYP450 3A4 Substrate | Substrate | Substrate | Substrate |
| 10. | CYP450 1A2 Inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor |
| 11. | CYP450 2C9 Inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor |
| 12. | CYP450 2D6 Inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor |
| 13. | CYP450 2C19 Inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor |
| 14. | CYP450 3A4 Inhibitor | Inhibitor | Inhibitor | Inhibitor |
| Toxicity | ||||
| 15. | AMES Toxicity | Non AMES toxic | Non AMES toxic | Non AMES toxic |
| 16. | Carcinogens | Non-carcinogens | Non-carcinogens | Non-carcinogens |
The compounds ZINC000150588351, ZINC000064540179 and ZINC000137700912 possess the human intestinal absorption capacity and lack the blood brain barrier and Caco2 (cell lines obtained from human colorectal carcinoma, used to find oral absorption) permeability. The Permeability glycoprotein (P-glycoprotein), a transmembrane protein involved in drug efflux from cells, shows non-substrate and non-inhibitor property for ZINC000064540179, ZINC000137700912 compounds and substrate, inhibitor property for ZINC000150588351. These three compounds have shown non-inhibitor property for drug clearance (Renal Organic Cation Transporter), also non-substrate status for the cytochrome P450 (CYP) isoforms CYP450 2C9 & CYP450 2D6, Non-inhibitor status for CYP450 2C9 & CYP450 2D6. These compounds are non-mutagenic, which has reported negative results for AMES toxicity. These three compounds are considered as the potential leads against the SARS-CoV-2 S D614G variant.
Conclusion
The outburst of the COVID-19 has challenged the public health, medical and economic infrastructure. In our current study, to screen active leads against the highly infectious, more stable pathogenic SARS-CoV-2 S D614G variant, we have employed structure-based virtual screening. We have subjected a total of 500 small molecules to the virtual screening process and found compounds ZINC000150588351, ZINC000064540179 and ZINC000137700912, which have high binding energy with highly infectious SARS-CoV-2 S D614G viral protein. These compounds can be further subjected to in-vitro and in-vivo studies to identify their antiviral activity. Our findings will be helpful for the researchers for further design and development of antiviral drugs against COVID-19.
Acknowledgment
KM, JMA and SKM would like to thank the management of PMCHRI for providing the necessary facilities to carry out this research work.
Conflict of interest
The authors declare that there is no conflict of interest.
Funding Sources
There is no funding source.
References
- Romeo A, Iacovelli F and Falconi M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res., 2020; DOI: 10.1016/j.virusres.2020.198068.
CrossRef - Zhou P, Yang X. L, Wang X. G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 2020; 579: 270-273.
CrossRef - Li H, Liu S. M, Yu X. H, Tang S. L and Tang C. K. Coronavirus disease 2019 (COVID-19): current status and future perspectives. Int. J. Antimicrob. Agents, 2020; 55: 105951.
CrossRef - Huang Y, Yang C, Xu X. F, Xu W and Liu S. W. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin., 2020; 41: 1141-1149.
CrossRef - Chan, J. F. W, Yao Y and Yeung M. L, et al. Treatment with lopinavir/ritonavir or interferon-β1b improves outcome of MERSCoV infection in a nonhuman primate model of common marmoset. Infect. Dis., 2015; 212: 1904-1913.
CrossRef - Tiwari V, Beer J.C, Sankaranarayanan N.V, Swanson-Mungerson M and Desai U.R. Discovering small-molecule therapeutics against SARS-CoV-2. Drug Discov. Today, 2020; 25: 1535-1544.
CrossRef - Walls A. C, Tortorici M. A, Frenz B, et al. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Struct. Mol. Biol., 2016; 23: 899-905.
CrossRef - Bosch B. J, Bartelink W, Rottier P. J and Cathepsin L. Functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. Virol., 2008; 82: 8887-8890.
CrossRef - Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun., 2020; 11: 1620.
CrossRef - Li W, Moore M. J, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003; 426: 450-454.
CrossRef - Watanabe R, Matsuyama S, Shirato K, Maejima M, Fukushi S, Morikawa S, Taguchi F. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved S protein as revealed by pseudotype virus bearing cleaved S protein. J. Virol. 2008; 82: 11985-11991.
CrossRef - Zhang L, Jackson C. B, Mou H, et al. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. BioRxiv, 2020, DOI: 1101/2020.06.12.148726.
CrossRef - Ogawa J, Zhu W, Tonnu N, Singer O, Hunter T, Ryan A. L, Pao G. M. The D614G mutation in the SARS-CoV2 Spike protein increases infectivity in an ACE2 receptor dependent manner. BioRxiv, 2020, DOI: 10.1101/2020.07.21.214932.
CrossRef - Yurkovetskiy L, Wang X, Pascal K. E, et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell. 2020 Sep 15:S0092-8674(20)31229-0.
CrossRef - Al-Tawfiq, J. A, Momattin H, Dib J and Memish Z. A. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. J. Infect. Dis. 2014; 20, 42–46.
- Wang Y, Zhang D, Du P., et al. Articles Remdesivir in adults with severe COVID-19: a randomised, doubleblind, placebo-controlled, multicentre trial. Lancet, 2020; 6736, 1–10.
- Berman H. M, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res., 2000; 28: 235-242.
CrossRef - Webb B and Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinformatics, 2016; 54: 5.6.1-5.6.37.
CrossRef - Morris G. M, Huey R, Lindstrom W, Sanner M. F, Belew R. K, Goodsell D. S, Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem., 2009; 30: 2785-2791.
CrossRef - Wiederstein, M and Sippl, M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res., 2007; 35: W407-410.
CrossRef - Pires D. E, Ascher D. B, Blundell T. L. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res., 2014; 42: W314-319.
CrossRef - Seifi M, Walter M. A. Accurate prediction of functional, structural, and stability changes in PITX2 mutations using in silico bioinformatics algorithms. PLoS One, 2018; 13: e0195971.
CrossRef - Irwin J. J and Shoichet B. K. ZINC–a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005; 45: 177-182.
CrossRef - Yang H, Lou C, Sun L, et al. AdmetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2019; 35: 1067-1069.
CrossRef - Hsu K.C, Chen Y.F, Lin S.R, Yang J.M. iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinformatics. 2011; 12 Suppl 1(Suppl 1): S33.
CrossRef - Bell A. M, Wagner J. L, Barber K. E, Stover K. R. Elbasvir/Grazoprevir: A Review of the Latest Agent in the Fight against Hepatitis C. Int. J. Hepatol., 2016; 2016: 3852126.
CrossRef - Bhatia H. K, Singh H, Grewal N, Natt N. K. Sofosbuvir: A novel treatment option for chronic hepatitis C infection. J. Pharmacol. Pharmacother., 2014; 5: 278-284.
CrossRef

