
Manuscript accepted on :08-12-2021
Published online on: 27-12-2021
Plagiarism Check: Yes
Reviewed by: Dr. Mostafa Abdulaal
Second Review by: Dr. Ismail Ismail Abdullahi
Final Approval by: Dr. Fai Poon
Suliman A. Alderhami1 , Khaled A. Abdelshafeek1,2,*, Walid E. Abdallah2 , Ahmed A. Elhenawy1,3 and AbdulAziz Ali Alomari1
, Khaled A. Abdelshafeek1,2,*, Walid E. Abdallah2 , Ahmed A. Elhenawy1,3 and AbdulAziz Ali Alomari1
1Department of chemistry, faculty of sciences and arts, Mukhwah , Albaha university, Albaha, Saudi Arabia
2Department chemistry of medicinal plants, Pharmaceutical Industries Div., National Research Centre, 33 El Buhouth St.(Former El Tahrir St.), 12622-Dokki, Giza, Egypt.
3Chemistry Department, Faculty of Science, Al-Azhar University (Boys Branch),Nasr City, Cairo, Egypt.
Corresponding Author E-mail: khabdelhady@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/2302
Abstract
Glucosinolates (Gls) are natural bioactive compounds that form metabolites called isothiocyanates (ITC) which have various therapeutic effects. This study aimed to isolate the glucosinolates of Carrichtera annua L.(DC) (CA) and Farsetia aegyptia Turra (FA) belonging to the Crucifereae family. Total Gls were isolated from the aqueous methanolic extract of the plants and further purified using an acidic aluminum oxide column. Some of the obtained Gls were identified via spectroscopic methods (UV, NMR, and MS) and the rest were hydrolyzed by myrosinase to the corresponding isothiocyanates (ITC) for identification by GC/MS. Only one Gls was identified in CA as 4-methylthio-3-butenyl Gls (MTBG) in addition to 6-methyl sulfonylhexyl isothiocyanates (ITC), while 6-methyl sulfonyl-6-hydroxy hexyl ITC, 4-pentenyl ITC, 3-methylthio propyl ITC, 5-hydroxy pentyl ITC and 4-methylsulphinyl butyl ITC were identified in FA. The Gls demonstrated high binding activity to α-glucosidase and amylase, good pharmacokinetic characteristics, and exerted no carcinogenetic effects.
Keywords
Crucifereae; Carrichtera annua; Docking; Farsetia aegyptia; Glucosinolates and Isothiocyanates; GC/MS
Download this article as:| Copy the following to cite this article: Alderhami S. A, Abdelshafeek K. A, Abdallah W. E, Elhenawy A. A, Alomari A. A. Glucosinolates with Their Hydrolysis Products from Two Cruciferous Plants with Study of Antidiabetic Activity Based on Molecular Docking. Biomed Pharmacol J 2021;14(4). |
| Copy the following to cite this URL: Alderhami S. A, Abdelshafeek K. A, Abdallah W. E, Elhenawy A. A, Alomari A. A. Glucosinolates with Their Hydrolysis Products from Two Cruciferous Plants with Study of Antidiabetic Activity Based on Molecular Docking. Biomed Pharmacol J 2021;14(4). Available from: https://bit.ly/3Jgyppj |
Introduction
Many plants of the Cruciferae (formerly Brassicaceae) family contain glucosinolates which are precursors of strongly odorous pungent isothiocyanates formed by the action of myrosinase on glucosinolate when the plant tissues are ruptured1 . The Cruciferae family is considered one of the largest angiosperm families and includes many economically important plants, such as salad vegetables and crop species. These plants have many biological activities, with anticancer, antibacterial, antifungal, antirheumatic, and antidiabetic properties2. Carrichtera and Farsetia are two genera belonging to the Crucifereae family. In Egypt, there is only one Carrichtera species, C. annua L.(DC) grown in the Sinai peninsula, especially in the Elarish region, while the Farsetia genus is represented by three species, of which, F. aegyptia Turra. is more popular and grown in many localities throughout Egypt3. Both plants have been used by the native Bedouins for their antidiabetic and antispasmodic effects. Moreover, F. aegyptia is used to relieve rheumatic pains and taken as a cooling medicine after pounding. F. aegyptia is also prepared as a decoction for toothache, gingivitis, and sore eyes, and mixed with alum for use as a mouth rinse and disinfectant2-4.
Glucosinolates (Gls) contain a thioglucosidic bond between the carbon of a sulfonated oxime and a side chain (R group) which may be aliphatic (e.g. alkyl, alkenyl, hydroxy alkenyl, thioalkyl), aliphatic, aromatic (eg. benzyl, substituted benzyl), or heterocyclic (e.g.indolyl) as shown in chart 1 [3]3. Gls and their hydrolysis products (isothiocyanates, ITC) have various biological activities with anticancer, antibacterial, antifungal, antioxidative, and allelopathic properties. ITCs such as sulforaphane, iberin, phenylethyl, and propenyl derived from glucoraphanin, glucoiberin, gluconasturtiin, and sinigrin induce phase-2 enzymes promoting antiproliferative activity5-8.
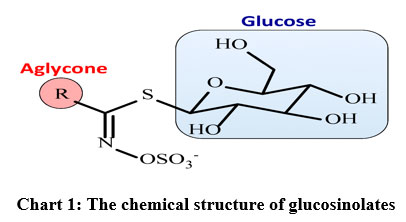 |
Chart 1: The chemical structure of glucosinolates |
The glucosinolates and their hydrolysis products in suspension culture following elicitation of F. aegyptia were found to contain glucotropaeolin (bezyl Gls) and gucocheirolin (3-methylsulfonylpropyl-Gls), isobutyl Gls and gluconasturtiin in addition to twenty-two hydrolysis products9-10. Total glucosinolates of C. annua and F. aegyptia exhibit strong antifeedant effects against the 4th instar larvae of the Egyptian cotton leaf worm, S. littoralis4.
Diabetes mellitus is a chronic endocrine disorder that affects the metabolism of carbohydrates, proteins, fat, electrolytes, and water. It is characterized by hyperglycemia in which blood sugar levels are elevated because the pancreas does not produce enough insulin or cells do not respond to the produced insulin11. Therefore, a therapeutic approach to treat diabetes is to decrease postprandial hyperglycemia12, which can be achieved by the inhibition of carbohydrate hydrolyzing enzymes like α-amylase and α-glucosidase, important enzymes involved in the digestion of carbohydrates.
Docking and DFT simulations are powerful computational tools that have been proven to be inexpensive and efficient approaches for understanding fundamental characters of biomolecules13. DFT calculations provide information regarding important topographic and molecular characteristics such as optimization geometry, charge-transformation, global and local reactivities14-16. Molecular docking studies explore the possible binding mode of the compound via its target protein, predicting the ligand-receptor orientation that is useful to guide improved compound features for drug design17. The present study isolated and identified glucosinolates and their hydrolysis products of C. annua and F. aegyptia, combining DFT, ADMET, and molecular docking analysis to investigate the potential antidiabetic properties.
Material and methods
Plant Materials
The aerial parts of both plants (Carrichtera annua (L.) DC. and Farsetia aegyptia Turra) were collected from Alariesh airport region (North Saini governate) and wadi Hagol (at Cairo-Sues Road, 30km), respectively, through the flowering stage at the end of the winter season (25th March). The plants were kindly, identified by Prof. Ibrahim Elgarf, professor of taxonomy at the Botany Dept., Faculty of Science, Cairo University, and the voucher specimens were deposited at NRC herbarium. Each plant was air-dried and ground to a fine powder.
Instruments and Reagents
1H and 13C NMR spectra were measured using Joel JNM EX270FTNMR system at 270 MHz for 1H and 70 MHz for 13C at the central lab-NRC, Cairo, Egypt. The sample was dissolved in DMSO and the chemical shifts are given in δ (ppm) relative to tetramethylsilane (Me4Si). The UV spectra were recorded on a Shimadzu model UV-240 and a 2401 PC spectrophotometer (Shimadzu Inc., Tokyo, Japan) in methanol in the range of 200-450 nm.
Conditions for GC/MS analysis of the isothiocyanates
Apparatus
GC/MS Finning an SSQ7000, Digital DEC3000 with column: DB5 capillary (I.d.0.25mm), ionization mode: EI at 70ev, temperature program starting at: 50˚C up to 250 ˚C with a rate of 4 ˚C/min., Detector: MS, sample volume 2 l and the mass from 40-400.
modified silver nitrate reagent12 in which the Gls. appears as brownish spots on a yellowish background.
Solvent systems: S1, Butanol: acetic acid : water 4:1:5 upper layer (which contain the organic solvent saturated with water) and S2, Butanol: ethanol: water 4:1:3. Acidic aluminium oxide (anionotropic 1.5 kg, activity grade I, type WA-I acidic, Sigma chemical Co.), Cellulose for column (Merck, 64271 Darmstadt, Germany) and DEAE Sephadex A-25 (Germany). myrosinase enzyme (Thioglucosidase, Sigma, T – 4528).
Extraction and Isolation of Glucosinolates
About 2.0 kg of air-dried powdered herb of both C. annua and F. aegyptia were defatted, separately, with petroleum ether (br. 40-60°C) in a Soxhlet apparatus for three days (operated 12 hrs. daily). The defatted powders were air -dried and extracted (separately) with methyl alcohol (80%, 3x3L, 3 days each), the combined methanol extract of each plant was evaporated in vacuo at 40 ºC and the residue was dissolved in hot distilled water (750 ml), left in the refrigerator for 24 hrs, and filtered the gummy ppt. The aqueous filtrates were allowed to pass slowly through an acidic aluminium oxide column (Aluminium oxide, for chromatography, acidic, Brockmann I, 50-200 µm, 60A, Acros Organics, Fluca, 7.5×60) to remove the coloring matter, then the total Gls were eluted with potassium sulfate solution (2%, 2.5L) until the brown zone (containing the total glucosinolates) reached the bottom of the column, and the eluate containing the brown zone was evaporated under reduced pressure till dryness. The residue for each plant was dissolved with hot methanol, filtered through a central glass funnel (G3) and evaporated in vacuo at 40°C till dryness to leave a faint brownish residue (2.1 g for CA and 500 mg for FA)
Purification of Gls
The obtained brownish residue was further purified using a cellulose column eluted with the upper layer of the solvent S2, after the complete elution, the eluate was concentrated and subjected to preparative paper chromatography (PPC) using chromatographic paper Whatmann 3 mm developed in the upper layer of solvent S1 (two runs) by applying the descending technique. Two main zones were determined with a pencil cut into small pieces and eluted with methanol (90%). Each zone was further purified over a DEAE Sephadex A-25 column eluted with 0.5% ammonium acetate soln., to afford two compounds (CA1 and CA2, Rb 0.52 and 0.65, relative to benzyl Gls, 17mg and 3mg, respectively).
Enzymatic hydrolysis of Gls
Due to the small amounts of the compound A2 and the residue of total Gls from FA, so compound A2 and the total Gls from FA were subjected, separately, to enzymatic hydrolysis using a myrosinase enzyme. The aqueous acidic solution after separation of the aglucones was neutralized with barium carbonate and filtered. The clear filtrate was evaporated till dryness. The residue was dissolved in 10% isopropanol and subjected to paper chromatography using ethyl acetate: pyridine: water (12: 5: 4) as the developing solvent and different authentic sugars. The chromatogram was visualized by spraying with aniline phthalate, and heated at 105 °C for a few minutes. Only glucose was detected as a sugar and the presence of sulfate ions in the aqueous solution was detected by the addition of a few drops of barium chloride soln., where a white ppt. was noticed, proving the presence of sulphate ions.
Also, the total Gls. were extracted from F. aegyptia using the same method, which were purified using PPC with different solvents and repeated Sephadex LH-20 columns to afford five compounds in small amounts, so they were subjected to enzymatic hydrolysis as before, and the hydrolysis products were extracted with ether and identified by GC/MS. The aqueous layer was found to contain glucose and sulphate ions.
Computational study
Preparation of the Small Molecule
The target compound was built and minimized using the PM3 semi-empirical Hamiltonian molecular orbital calculation MOPAC16 package 32, then employing the density function theory in the Gaussian 09 program package28 with the Becke3-Lee-Yang-parr (B3LYP) level using a 6-311G* basis, as implemented in the MOE 2015 package17. The optimization Geometry for molecular structures was applied to improve the knowledge of chemical structures. Our compounds were introduced into the binding sites according to the published crystal structures. The global chemical reactivity descriptors for molecules were computed, like: S; softness (measures the stability of molecules and chemical reactivity with direct proportional), η; hardness (reciprocal of softness), µ; chemical potential, χ; electronegativity strength catching electrons, the μ−; electron donating power, μ+; potency for catching the electron, ω−; electro donating capacity, ω+; electro accepting capacity, ω±; net electrophilicity (measuring the relative power between electron acceptance and electron donatation) 15, wi; an electrophilicity index in a ground state (determining the decreasing energy obtained from the maximal electrons’ current movement between the donor and acceptor media), wiVS; and the electrophilicity index in the valance state. These parameters are represented in terms I; ionization potential, and A; electron affinity15, as the previous terms represented.
Selection of protein structures
A docking experiment was carried out for the target active site into α-glucosidase (PDB: 4yvx) and β-amylase (PDB: 4gqr) using MOE 201517. The errors of the active sites were corrected by the structure preparation process in MOE. After the correction, hydrogens were added and the partial charges (Amber12:EHT) were calculated. Energy minimization (AMBER12:EHT, the root mean square gradient: 0.100) was performed.
Binding site analysis
The binding site of each receptor was identified through the MOE Site Finder program, which uses a geometric approach to calculate the putative binding sites in a protein, starting from its tridimensional structure. This method is not based on energy models, but only on alpha spheres, which are a generalization of convex hulls. The prediction of the binding sites, performed by the MOE Site Finder module, confirmed the binding sites defined by the co-crystallized ligands in the holo-forms of the investigated proteins.
MOE Stepwise Docking Method
The crystal structures of the enzymes were obtained. Water and the inhibitors’ molecule were removed, and hydrogen atoms were added. The parameters and charges were assigned with the MMFF94x force field. After that, the alpha-site spheres were generated using the site finder module of the MOE. The optimized 3D structures of the molecules were subjected to generate different poses of ligands using the triangular matcher placement method, which generates poses by aligning the ligand triplets of the atoms with triplets of the alpha spheres represented at the receptor site points. A random triplet of the alpha sphere center was used to determine the pose during each iteration. The pose generated was rescored using the London dG. scoring function. The poses generated were refined with the MMFF94x forcefield; also, the solvation effects were treated. The Born solvation model (GB/VI) was used to calculate the final energy, and the finally assigned poses were assigned a score based on the free energy in Kcal/mol.
ADMET predictions
The ADMET in silico profile was applied using “MOE” and “admetSAR” tools to predict the pharmacokinetic and ADMET characters (absorption, distribution, metabolism, excretion, and toxicity).
Results and discussion
Isolation and identification of Gls
The total Gls of both plants were isolated and further purified yielding two compounds (CA1 and CA2) from CA and five compounds (FA1-FA5) from FA as follows:
CA1
CA1-4-methylthio-3-butenyl Gls: This off-white amorphous powder appeared as a brown spot (Rb = 0.52) after spraying with a modified AgNO3 reagent. The UV absorption spectrum of the compound in methanol had a λmax .at 208 nm which shifted to 217 nm on the addition of NaOH. When silver nitrate solution (1%) was added a new maximum appeared at 260 nm in addition to the first peak at 215 nm with an increase in intensity indicating the glucosinolate nature of the compound18.
The 1H-NMR spectrum (DMSO) showed signals at in ppm 2.13 (3H, s, CH3-S), 2.35 (2H, t, H-1), 2.7 (2H, q, H-2), 4.75 (1H, d, J=8.2Hz with a small t, H-3), 5.8 (1H, d, J=8.2Hz, H-4), the anomeric proton of glucose was displayed as a doublet at 5.1ppm. The complex group of signals between 3.1 and 3.85 is characteristic for the rest proton of the glucose moiety. The 13C-NMR spectrum gave the anomeric carbon of the glucose unit at 82.16, the two carbon atoms (C-3 and C-4) of the double bond appeared at 137 and 115.6 ppm respectively, and the central carbon of the Gls was assigned at 156.21 ppm. The other data were summarized in Table-1.
Table 1: 13C-nmr data of compound CA1.
| Carbon no. | δin ppm |
| C1 | 31.75 |
| C2 | 30.44 |
| C3 | 137.01 |
| C4 | 115.6 |
| S-C5 | 24.35 |
| C=N | 156.21 |
| C1′ | 82.16 |
| C2′ | 72.94 |
| C3′ | 78.15 |
| C4′ | 69.79 |
| C5′ | 81.15 |
| C6′ | 60.93 |
These findings are in line with a report by Manuela et al.11. The GC/MS of the isolated aglucone of this compound after the enzymatic hydrolysis identified the corresponding ITC with the following fragmentation pattern: the mass spectrum displayed a mass to charge ratio (m/z) of 159 due to the molecular ion peak M+ which corresponded to the molecular formula C5H7S2N and fragment ion peaks at m/z 144 (M+ – CH3), 101 (M+ – NCS) and 73 (M+ – CH2NCS). By comparing these data with the reported data, the compound CA1 was identified as 4-methylthio-3-butenyl glucosinolate.
CA2
CA2 – 6-methylsulfonylhexyl Gls: This compound was isolated in a small amount (~ 3 mg) as a white-yellowish powder which was not sufficient for NMR measurements, so was subjected to enzymatic hydrolysis. The resultant ITC was identified as follows: the mass spectrum exhibited M+ at m/z of 221 (20.5%) which fits the molecular formula C8H15O2NS2. The other fragments were at m/z = 220 (M+-1, 23%), 205 (M+– CH3, 100%), 163 (M+– NCS, 40%) and 142 (M+– CH3SO2, 35%). This fragmentation pattern confirmed the identification of 6-methylsulfonylhexyl ITC [CH3SO2-CH2-(CH2)4-CH2-NCS] and accordingly the compound was identified as 6-methylsulfonylhexyl Gls. This is the first report of the isolation of these compounds from C. annua grown in Egypt.
The GC/MS data of the Gls confirmed that the FA hydrolysis products matched previously published data18-19 and included only isothiocyanates which were identified in an increasing retention manner as follows:
FA1
FA1 – 6-methylsulfonyl6-hydroxy-hexyl ITC: Rt. 15.3 min, the mass spectrum displayed a molecular ion peak M+ at m/z (% relative abundance) of 237 (5%) corresponding to the molecular formula C8H15O3NS2. Other fragments at 236 (M+ -1, 12.3%), 221 (M+ – CH4, 28.6%), 204 (M+– SH, 17.5%), 180 (M+– NCS, 74%), 165 (M+– CH2NCS, 100%), 137 (M+– (CH2)3NCS, 32%), 123 (M+– (CH2)4NCS, 12.5%), 109 (M+– (CH2)5NCS, 18%) and 91 (M+– [H2O +(CH2)5NCS], 11.8%) or (M+-[CH3SO2-CH-1]). The latter two fragments (m/z 91 and 109) showed the presence of an OH group at C-6 and the fragmentation pattern indicated 6-methylsulfonyl–6-hydroxy hexyl ITC [CH3SO2-CH(OH)-(CH2)4-CH2– NCS] (Chart 2).
FA2
FA2 – 4-pentenyl ITC: Rt. 14.4 min, the molecular ion peak M+ was present at m/z=127 (14.3%) which corresponds to the molecular formula C6H9NS. Other important fragments at m/z 100 (M+– (CH2=CH+, 18%), 85 (M+-(CH2)3, 76%) and 57 (M+– (CH2=CH-(CH2)3, 100%) confirmed the compound as 4-pentenyl ITC CH2=CH-(CH2)3-NCS (Chart 2).
FA3
FA3 – 3-Methylthio propyl ITC: Rt. 16.9 min, the mass spectrum gave M+ at m/z of 147(12%) which fits with the molecular formula C5H9NS2. The peaks at m/z 101 (M+- CH3S, 100%), 86 (M+- CH3S-CH2, 2%), 72 (M+- CH3S-CH2-CH2, 50%), and 61 (M+- -CH2-CH2-NCS, 44%) confirmed the presence of a methylthio unit attached to a propyl group and the compound was identified as 3-methylthio propyl ITC CH3S-CH2-CH2-CH2-NCS (Chart 2).
FA4
FA4 – 5-hydroxypentyl ITC: Rt. 19.2 min, the mass spectrum of this compound showed M+ at m/z =145 (3%) corresponding to the molecular formula C6H11ONS. The fragments at 101 (M+ -(CH2-OH), 22.5%), 87 (M+ – NCS, 20.6%), 73 (M+– CH2-NCS, 67.8%) and 59 (M+ – [CH2-(OH)CH2CH2, 100%) confirmed the identification of 5-hydroxypentyl ITC [CH2 (OH)-(CH2)3-CH2– NCS] (Chart 2).
FA5
FA5 – sulphoraphane (4-methylsulphinyl butyl ITC): Rt. 28.7 min, the MS of the compound revealed the presence of M+ at m/z= 177 corresponding to C6H11ONS2. The fragmentation gave rise to 114 (M+ – CH3SO-, 6%), 86 (M+ – CH3SO-CH2, 5%) and 72 (M+ – CH2NCS, 100%), identifying the compound as 4-methylsulphinyl butyl ITC, CH3SO-(CH2)4NCS (Chart 2).
The variation in the glucosinolate hydrolysis products between previous studies and the present work may be due to geographical and seasonal differences, environment, soil type, stress, and plant part examined, or due to the differences in the experimental conditions.
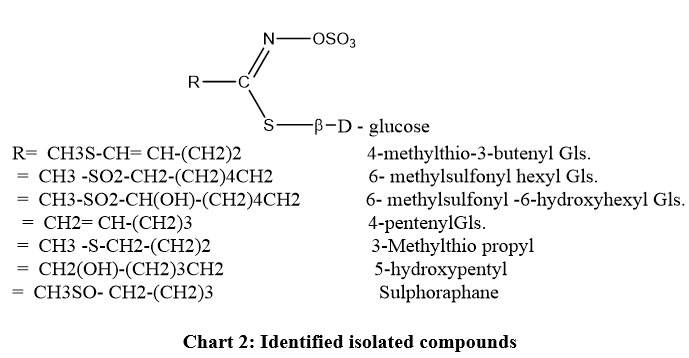 |
Chart 2: Identified isolated compounds |
Computational studies
Frontier orbital analysis
“FMOs” frontier molecular orbitals through two types HOMO (donating electron) and LUMO (accepting electron) are crucial orbitals for molecules, displaying the binding properties of biomolecules with a receptor. The FMOs were characterized by the chemical reactivity and kinetic stability of the molecule19-20.
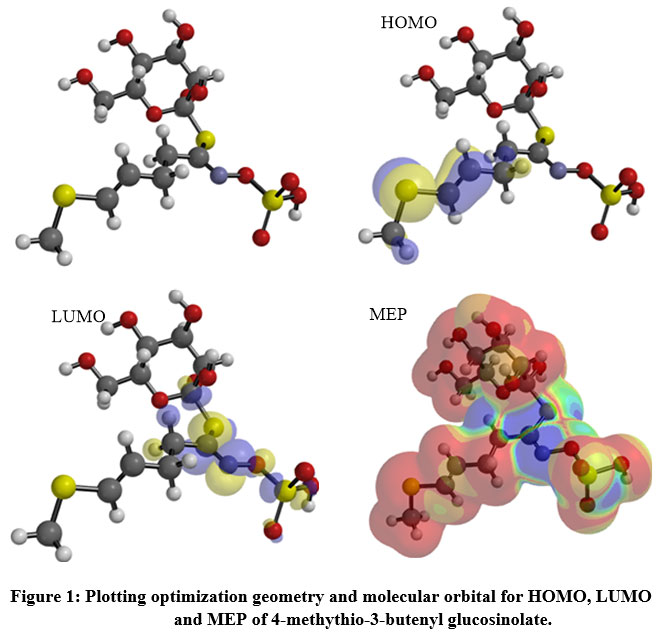 |
Figure 1: Plotting optimization geometry and molecular orbital for HOMO, LUMO and MEP of 4-methythio-3-butenyl glucosinolate. |
The HOMO was localized over the methyl(prop-1-en-1-yl)sulfane region (Figure 1), while the sulfanecarbaldehyde O-((oxidaneyl)dioxo-l6-sulfanyl) oxime fragment was covered by the LUMO zone, thus, electron transition (HOMO→LUMO) from sulfonyl to sulfonyl groups. The low DG enhances the interaction between the HOMO ligand and the LUMO receptor (Table 2) with an inversed mode 20. The charge transfer was represented by the reactivity index term “DNmax”, which was restricted by the stabilization energy during the system gaining charge from the biological media (Table 2). The low softness value may be combined with a high potential against a biological environment (Table 2). The ligand had a high electrophilicity value “ω±= 6.741”, increasing the electron-donating power (µ-=7.02ev) with increasing donating capacity (ω– = 6.74ev), which may increase the attacking power against a polar residue in the receptor, thereby increasing the potency.
Table 2: Calculated energetic global reactivity parameters 4-methythio-3-butenyl gluconate at DFT with a B3LYP\6-311G* Basics sets
| E | -1554.78 | E: The total energy (Kcal/mol)., HOMO: Highest Occupied Molecular Orbital (eV), LUMO: Lowest Occupied Molecular Orbital (eV), ΔG: difference between HOMO and LUMO energy levels(eV), I: Ionization potential, A; electron affinity; η: Hardness(eV), S: Softness(eV), χ : Electronegativity (eV; µ+: electron accepting chemical potentials , µ-: electron donating chemical potentials, µ+: chemical potential(eV), ω+: electron accepting capacity, ω-: Electrodonating capacity; ω±: :Electrophilicity index in valance state (eV); ΔNmax: maximum number of electrons transfer. |
| HOMO | -9.23 | |
| LUMO | -1.39 | |
| ΔG | -7.84 | |
| I | 9.23 | |
| A | 1.39 | |
| η | 3.92 | |
| S | 0.255 | |
| χ | -5.31 | |
| µ + | -3.35 | |
| µ – | -7.027 | |
| ω – | 6.741 | |
| ω + | 3.106 | |
| ω ± | 9.847 | |
| DNmax | 1.354 |
Molecular electrostatic potential (MEP)
The repulsive and attractive forces were determined by the molecular electrostatic potential (MEP) for 4-methylthio-3-butenylglucosinolate (Figure 1). Orange, yellow, red colors depict the high electron density area, while the blue color represents the positive potential, and the green color represents the intermediate potential. The negative charge capped most of the molecule area, while the formaldhydoxime area was covered by a positive charge. The variation in color of the MEP surface is associated with a difference in the electrostatic potential. This force is accountable for the interaction of the substrate with the receptor binding site, hence promoting the electrostatic bond between the substrate and receptor21.
Docking studies
The The docking study targeted α-glucosidase and β-amylase to examine the mode of action of 4-methylthio-3-butenylglucosinolate. The ligand-protein interaction was estimated based on the gold score function as implemented in MOE 2015.1022. All calculations are presented in Table 2. The crystal structures for α-glucosidase (PDB: 4yvx23) and α-amylase (PDB: 4gqr24) were obtained from the protein databank and they were complexed with glimipride and myricetin as reference drugs, respectively. The tested ligand docked into the active site of the two enzymes.
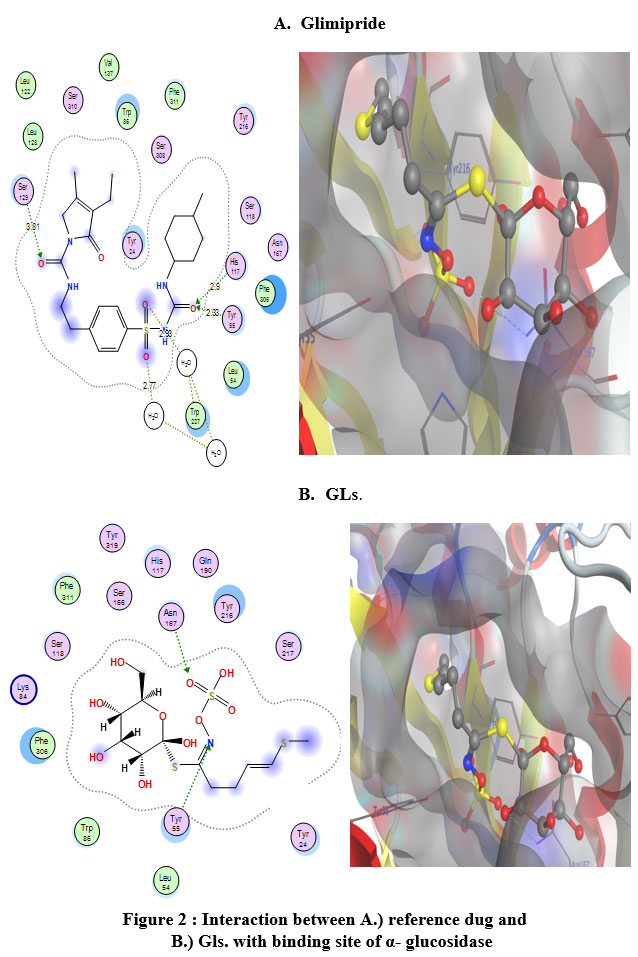 |
Figure 2 : Interaction between A.) reference dug and B.) Gls. with binding site of α- glucosidase |
The active site of these enzymes was defined to include residues within a 3.5 Å radius around the reference drug atoms. The molecular dynamic (MD) using an (MMFF94) force field to the 0.05 Kcal/mol gradient convergence range was applied to minimize the energy for the obtained ligand-enzyme complexes. The binding affinity of the ligands tested was determined by the highest MOE scoring function (Table 3).
Table 3: Docking energy scores (Kcal/mol) derived from the MOE for 4-methythio-3-butenyl gluconate.
| Enzyme | 4yvx | 4gqr | ||
| Glimipride | Tested Ligand | myricetin | Tested Ligand | |
| E_score1 | -7.75118 | -7.11842 | -101.157 | -131.364 |
| E.dGE | 1.62884 | 1.122716 | -5.19 | -7.209 |
| E_conf | -263.798 | 85.79013 | -101.987 | -117.44 |
| E_place | -49.6922 | -104.664 | -367.2 | -566.262 |
| E.Int. | -13.9234 | -14.7619 | -101.987 | -117.44 |
| E.H.B. | -42.0326 | -39.5142 | -37.74 | -56.14 |
| Eele | -7.75118 | -7.11842 | -3.07 | -3.6 |
| Evdw | -7.49118 | -7.28842 | 2.59 | 3.24 |
| RMS | 1.62884 | 1.122716 | 0.9652 | 0.865 |
E_score1; Initial free binding energy of the ligand from a given pose. Ed.G.: Final free binding energy of the ligand from a given pose, E_conf; Free binding energy of the ligand from a given conformer. E_place. Free binding energy of the ligand from a receptor. E.Int.: Affinity binding energy of ligand with receptor, H.B.: Hydrogen bonding energy between protein and ligand. Eele: Electrostatic interaction with the receptor, Evdw: Van der Waals energies between the ligand and the receptor. RMSD; The root mean square deviation of the pose of the docking pose compared to the co-crystal ligand position.
The premier ligands had a MOE score of -7.511 and -6.157 Kcal/mol, respectively, for α-glucosidase and β-amylase. In the α-glucosidase binding site, glimipride forms an important strong hydrogen bond with His117 and Tyr55, at the time this ligand forms p-p bonds with Tyr24 and Trp22. The analysis of the binding site of β-amylase-myricetin showed that the ligand interacted with an important amino acid backbone Asp197, which contacted 2-H-bonds. The tested compound exhibited a higher binding affinity with a MOE score of -6.611 Kcal/mol than the reference drug (-6.115 Kcal/mol.). This ligand formed an important hydrogen bond with Asp (Figure 3) and was arranged in a binding pocket in a parallel mode with Asp (Figure 3).
The hydrophilicity of the binding pocket indicates that the hydrophobicity and membrane permeability are important pharmacokinetic characteristics for absorption molecules in biological systems. These results demonstrate that the amino acid residues close to the reference molecules are mostly the same as those observed in the test compounds.
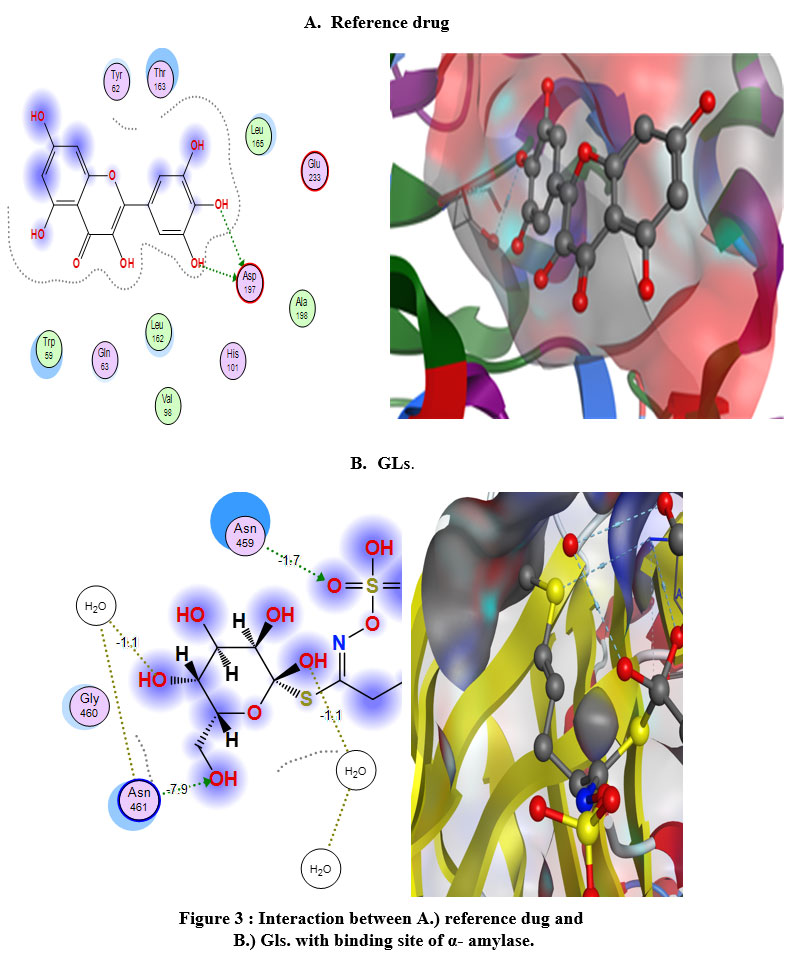 |
Figure 3 : Interaction between A.) reference dug and B.) Gls. with binding site of α- amylase. |
In silico Toxicological study
In silico pharmacokinetic Profile
The calculated descriptors for 4-methylthio-3-butenylglucosinolate and glimipride were calculated by MOE, SwissADME25, and the admet-SAR model26, as shown in Table 4. The physicochemical and ADME parameters for 4-methythio-3-butenyl glucosinolate glimipride (reference drug) indicate that this ligand is suitable for Lipinski’s rule with one violation related to molecular weight. Furthermore, when the Ghose Veber, Egan and Muegge rules were applied only glimipride qualified for the Muegge rule. Consequently, the test compound exhibited bioavailability scores of 0.11 and 0.55 with biodegradation values 0.6362 and 0.4523, confirming good oral bioavailability. The lead-likeness profile plotted the Bioavailability Radar planner for the test compounds (Figure 4) showing the relationship between polarity, size, lipophilicity, solubility, saturation, and flexibility20, with the optimal range for each parameter represented in pink. From Figure 4, 4-methythio-3-butenyl glucosinolate and glimipride exhibited deviation for polarity and flexibility, respectively.
Both compounds exhibited a high degree of saturation (Fraction Csp3 = 0.75,0.45, respectively), which pass the filter of Fraction Csp3< 0.2527. The solubility parameters are important for absorption, with 4-methythio-3-butenyl glucosinolate being strongly soluble in H2O when the ESOL topological model was applied with absorption% (%ABS=44.48)28, whereas glimipride showed poor solubility in this model (Table 4).
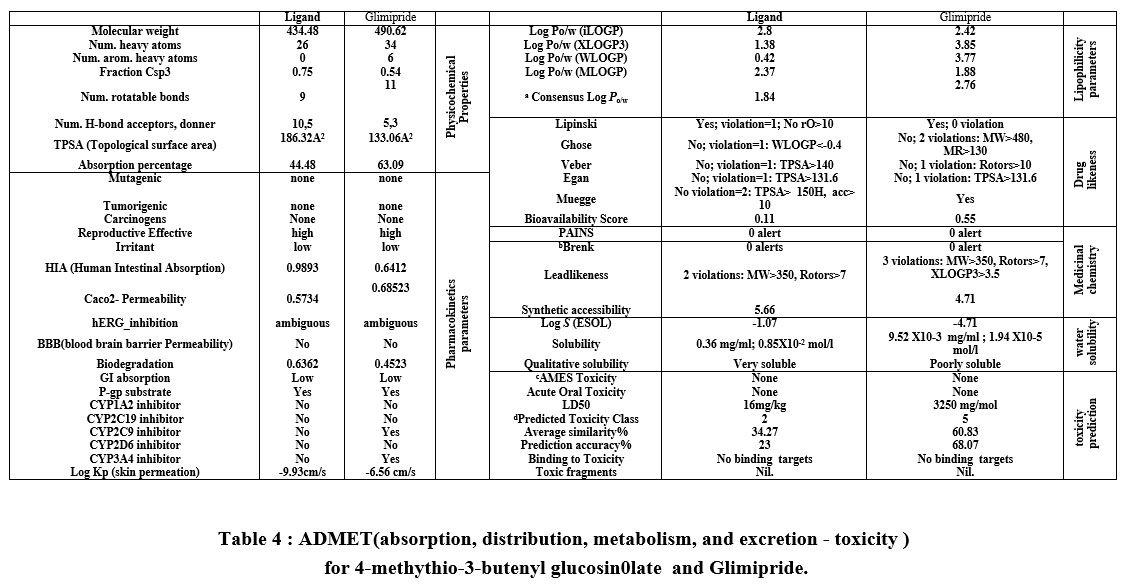 |
Table 4 : ADMET(absorption, distribution, metabolism, and excretion – toxicity ) for 4-methythio-3-butenyl glucosin0late and Glimipride. |
for 4-methythio-3-butenyl glucosin0late and Glimipride.
a Consensus Log Po/w: average four prediction of partition coefficient between n-octanol- water.
bhet-C-het_not_in_ring, imine_1, imine_2, oxygen-nitrogen_single_bond
c In vitro Ames test result for each TA100 strain (Metabolic activation by rat liver homogenate), TA100 strain (No metabolic activation), TA1535 strain (Metabolic activation by rat liver homogenate) and TA1535 strain (No metabolic activation)[9].
d Toxicity Class ranging from 1 to 6 according to the Global Harmony System (GHS) [8].
Also, the lipophilicity parameters in medicinal chemistry were incorporated as a lead-likeness filter to identify problematic fragments in bioactive molecules based on two structural alerts, PAINS and Brenk’s filters20. The tested compounds exhibited no structural alerts against PAINS and Brenk’s filters.
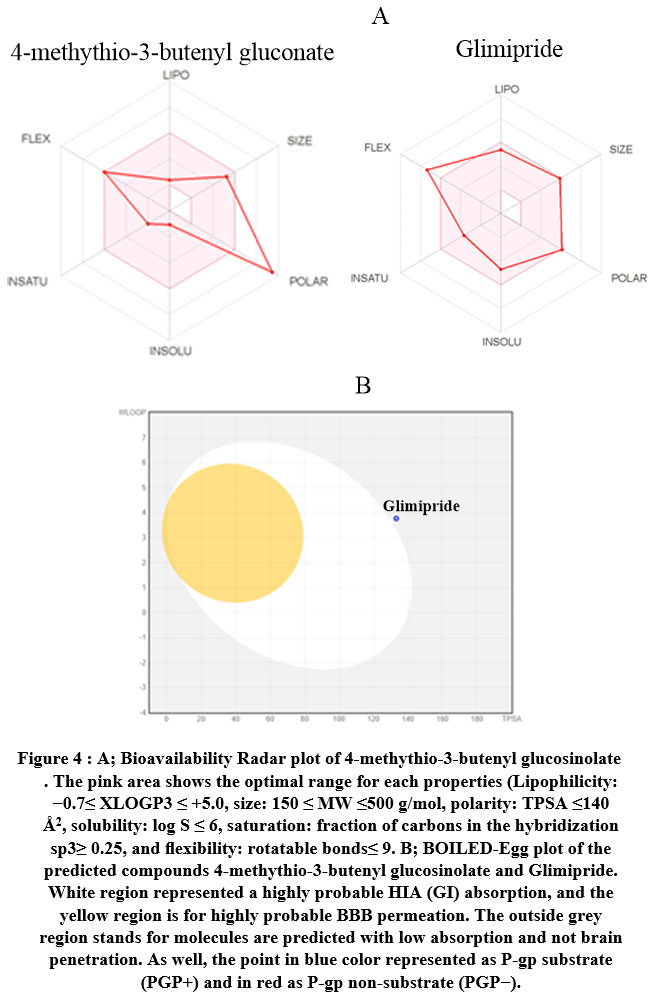 |
Figure 4 : A. Bioavailability Radar plot of 4-methythio-3-butenyl glucosinolate . The pink area shows the optimal range for each properties |
The mutagenic, tumorigenic, reproductive, and irritant properties and human intestinal absorption were investigated in silico using ADMET-SAR pharmacokinetic parameters29. Furthermore, the SVM algorithm was applied to identify whether the substrate or non-substrate could permeate the skin (Log Kp), Caco-2, blood-brain barrier (BBB), and p-glycoprotein (P-gp), as well as determine the inhibitory effect against the main cytochromes P450 isoenzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4). The results presented in Table 3 show that glimipride inhibited both CYP2C9 and CYP3A4 but the test ligand did not inhibit any P450 isoenzymes. The BOILED-Egg model represents the relationship for WLOGP vs TPSA in Figure 4, which demonstrated high BBB permeability, high GI absorption, high brain penetration, and ambiguous inhibition action against human Ether-à-go-go-Related Gene (hERG). Glimipride activated P-gp (multidrug resistance protein 1) but the test compound acted as a P-gp inhibitor. Furthermore, the skin permeability (Kp) results in Table 4 showed that glimipride has higher skin permeability than the test ligand. The tested compounds exhibited no carcinogenicity, mutagenicity, and tumorigenicity effects30 .
Oral toxicity prediction
The possible toxicity was predicted 31 by estimating the rodent oral toxicity based on data extracted from the Chemical European Biology Laboratory (ChEMBL) database 26. Also, the median lethal doses (LD50) were estimated in rodents, with the test ligand (LD50 = 16 mg/Kg) showing a lower value than the reference drug (LD50= 3250 mg/Kg). The calculated toxicity using this database depends on the highest endpoints including 33 models (Table 4). The calculated schematic is divided into several stages of toxicity, “toxicity, toxicological endpoints (mutagenicity, carcinotoxicity, organ toxicity (hepatotoxicity), cytotoxicity and immunotoxicity), toxicological pathways (AOPs) and toxicity targets thereby”25 providing information regarding the possible molecular mechanism as well as toxic response (Table 5). Glimipride was located in the lower toxicity class 5 than 4-methythio-3-butenyl glucosinolate in class (No. 4), with both compounds not exhibiting any toxic fragments without non-binding to any toxicity targets (Table 5).
Table 5: Toxicity Model Report for ligand which extracted from admet-SAR.
| Classification | Target | Shorthand | Prediction | Probability | Prediction | Probability |
| Glucosanate | Glimipride | |||||
| Organ toxicity | Hepatotoxicity | dili | Inactive | 0.62 | Inactive | 0.71 |
| Toxicity end points | Carcinogenicity | carcino | Inactive | 0.56 | Inactive | 0.72 |
| Toxicity end points | Immunotoxicity | immuno | Inactive | 0.84 | Inactive | 0.99 |
| Toxicity end points | Mutagenicity | mutagen | Inactive | 0.58 | Inactive | 0.76 |
| Toxicity end points | Cytotoxicity | cyto | Inactive | 0.7 | Inactive | 0.64 |
| Tox21-Nuclear receptor signaling pathways | Aryl hydrocarbon Receptor (AhR) | nr_ahr | Inactive | 0.91 | Inactive | 0.97 |
| Tox21-Nuclear receptor
signaling pathways |
Androgen Receptor (AR) | nr_ar | Inactive | 0.9 | Inactive | 0.98 |
| Tox21-Nuclear receptor
signaling pathways |
Androgen Receptor Ligand
Binding Domain (AR-LBD) |
nr_ar_lbd | Inactive | 0.96 | Inactive | 0.96 |
| Tox21-Nuclear receptor
signaling pathways |
Aromatase | nr_aromatase | Inactive | 0.92 | Inactive | 0.97 |
| Tox21-Nuclear receptor
signaling pathways |
Estrogen Receptor Alpha (ER) | nr_er | Inactive | 0.83 | Inactive | 0.96 |
| Tox21-Nuclear receptor
signaling pathways |
Estrogen Receptor Ligand
Binding Domain (ER-LBD) |
nr_er_lbd | Inactive | 0.91 | Inactive | 0.99 |
| Tox21-Nuclear receptor
signaling pathways |
Peroxisome Proliferator Activated
Receptor Gamma (PPAR-Gamma) |
nr_ppar_gamma | Inactive | 0.96 | Inactive | 0.95 |
| Tox21-Stress response
pathways |
Nuclear factor (erythroid-derived 2)-like 2/
antioxidant responsive element (nrf2/ARE) |
sr_are | Inactive | 0.96 | Inactive | 0.99 |
| Tox21-Stress response pathways | Heat shock factor response element (HSE) | sr_hse | Inactive | 0.96 | Inactive | 0.99 |
| Tox21-Stress response pathways | Mitochondrial Membrane Potential (MMP) | sr_mmp | Inactive | 0.92 | Inactive | 0.85 |
| Tox21-Stress response pathways | Phosphoprotein (Tumor Supressor) p53 | sr_p53 | Inactive | 0.93 | Inactive | 0.96 |
| Tox21-Stress response pathways | ATPase family AAA
domain-containing protein 5 (ATAD5) |
sr_atad5 | Inactive | 0.98 | Inactive | 0.99 |
In general, the tested compounds have good oral bioavailability, high ability BBB transport, and no marked health effects observed for rodent toxicity profiles.
Conclusion
The glucosinolates of Carrichtera annua and Farsetia aegyptia (Crucifereae family) were identified as 4-methylthio-3-butenyl Gls and 6-methyl sulfonylhexyl Gls from CA, 6-methylsulfonyl-6-hydroxy hexyl ITC, 4-pentenyl Gls, 3-methylthio propyl ITC, 5-hydroxy pentyl ITC and 4-methylsulphinyl butyl ITC from FA. Future experiments will be conducted to isolate a sufficient quantity of the glucosinolates of Farsetia aegyptia for identification and to investigate their antitumor activity. The obtained docking data showed that the ability of Gls to interact with α-glucosidase and amylase, thus this Gls may be a suitable inhibitor and used as an antidiabetic agent. The ADMET in silico showed that these compounds have good oral bioavailability and a high ability for BBB transport. Furthermore, these compounds had no observed carcinogenic and health effects.
Acknowledgment
there is no acknowledgment because the authors spent on the research (chemicals and analyses) from their own expense, therefore I asked Dr. sheng to reduce the publication fees and he promised me after the acceptance of the paper for pulication.
Contributions
Khaled.A.A.: conceived and designed the experiments. Walid.E.A: performed the experiments and wrote the paper. Ahmed.A.E.: have performed evaluation and validation of docking methods and scoring functions and drafted the manuscript. A.A.E. and K.A.A.: analyzed and interpreted the data. Khaled.A.A. and Suliman.A.A. .: outlined the research strategy and revised the manuscript, completed the literature search. and provided editorial support. All authors read and approved the final manuscript.
Conflict of interests
The authors declare that they have no competing interests.
Funding Sources
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- Rizk A. Phytochemistry of the Flora of Qatar.( 1986).
- Boulos L. Flora of egypt: Al Hadara Publishing Cairo; (2005).
- Abdelshafeek KA, ElMissiry MM, Hussiny HA, Seif MM. The Flavonoids and Anticomplement Activity of two Cruciferous Plants Growing in Egypt. . Int. J. Pharmacogn. Phytochem. Res. 8, 223–22, (2016)7.
- Bhandari SR, Jo JS, Lee JG. Comparison of Glucosinolate Profiles in Different Tissues of Nine Brassica Crops. Molecules. ;20(9):15827-41, (2015). Epub 2015/09/04. doi: 10.3390/molecules200915827. PubMed PMID: 26334264; PubMed Central PMCID: PMCPMC6331803.
CrossRef - Bellostas N, Sørensen JC, Sørensen H, Agriculture. Profiling glucosinolates in vegetative and reproductive tissues of four Brassica species of the U‐triangle for their biofumigation potential. J Journal of the Science of Food. ;87(8):1586-94, (2007).
CrossRef - Clarke DBJAM.Glucosinolates, structures and analysis in food.Analytical Metho;2(4):310-25, (2010).
CrossRef - Fahey JW, Zalcmann AT, Talalay PJP. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry ;56(1):5-51,(2001).
CrossRef - Al‐Gendy A, Lockwood GJF, GC–MS analysis of volatile hydrolysis products from glucosinolates in Farsetia aegyptia var. ovalis. flavor and fragranc J;18(2):148-52(2003).
CrossRef - Al-Gendy AA, Lockwood GB. Production of glucosinolate hydrolysis products in Farsetia aegyptia suspension cultures following elicitation. Fitoterapia;76(3-4):288-95, (2005). Epub 2005/05/12. doi: 10.1016/j.fitote.2005.03.013. PubMed PMID: 15885925.
CrossRef - Visentin M, Tava A, Iori R, Palmieri. Isolation and identification for trans-4-(methylthio)-3-butenyl glucosinolate from radish roots (Raphanus sativus L.). J. agricul. and food chem., ( 40(9):1687-91,(1992).
CrossRef - Nair SS, Kavrekar V, Mishra AJEJoEB. In vitro studies on alpha amylase and alpha glucosidase inhibitory activities of selected plant extracts. European Journal of Experimental Biology 3(1):128-32,(2013).
- Agarwal P, Gupta . Alpha-amylase inhibition can treat diabetes mellitus. RRJMHS;5:1-8, (2016).
- Elhenawy AA, Al-Harbi L, El-Gazzar M, Khowdiary MM, Moustfa A. Synthesis, molecular properties and comparative docking and QSAR of new 2-(7-hydroxy-2-oxo-2H-chromen-4-yl) acetic acid derivatives as possible anticancer agents. J Spectrochimica Acta Part A: Molecular Biomolecular Spectroscopy.;218:248-62, (2019).
CrossRef - Elgendy A, Nady H, El-Rabiei M, Elhenawy AAJRA. Understanding the adsorption performance of two glycine derivatives as novel and environmentally safe anti-corrosion agents for copper in chloride solutions: experimental, DFT, and MC studies. RSC Advance, 72:42120-3, (2019)1.
CrossRef - Ali IO, Hanafy AI, Salama TM, El-Nasser KS, El‐Henawy AA, Guma HA, et al. Synthesis, structural configuration and DFT molecular orbital studies of [Mn-2 [benzo [b] oxazole] acetonitrile] complexes encapsulated in ZSM-5: Direct synthesis of phenol by benzene hydroxylation. Microporous and Mesoporous Materials;262:35-48m (2018).
CrossRef - El-Henawy AA, Basta AH, El-Saied H, Abdel-Shakour EH, Hasanin M, El-Sheikh. Discovery potent of Bagasse (CMC-L-Phe) as bioactive material based on DFT calculations. HHJEJAC;13(5):1-16,(2018).
CrossRef - Kitchen DB, Decornez H, Furr JR, Bajorath JJNrDd. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov,3(11):935-49,(2004).
CrossRef - Luque FJ, López JM, Orozco M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theoretical Chemistry Accounts: Springer; 2000. p. 343-5.
CrossRef - Fukui K. Role of frontier orbitals in chemical reactions. Science.;218(4574):747-54, (1982). Epub 1982/11/19. doi: 10.1126/science.218.4574.747. PubMed PMID: 17771019.
CrossRef - Wildman SA, Crippen GM. Prediction of physicochemical parameters by atomic contributions. Journal of chemical information and computer sciences;39(5):868-73,(1999).
CrossRef - Elhenawy AA, Al-Harbi L, Moustafa GO, El-Gazzar M, Abdel-Rahman RF, Salim AEJDD, Development, et al. Synthesis, comparative docking, and pharmacological activity of naproxen amino acid derivatives as possible anti-inflammatory and analgesic agents. Drug Design, Development and Therap;13:1773-90, (2019).
CrossRef - Molecular Operating Environment (MOE) CCGU, 1010 handbook St. West, Suite 910, Montreal, QC, Canada, H3A 2R7, 2017.
- Corbeil CR, Williams CI, Labute PJJoc-amd. Variability in docking success rates due to dataset preparation. J Comput Aided Mol Des;26(6):775-86, (2012).
CrossRef - Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep.;7:42717, (2017). Epub 2017/03/04. doi: 10.1038/srep42717. PubMed PMID: 28256516; PubMed Central PMCID: PMCPMC5335600.
CrossRef - Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z, et al. admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics;35(6):1067-69,(2018).
CrossRef - Ritchie TJ, Ertl P, Lewis R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov Today.;16(1-2):65-72,(2011). Epub 2010/11/16. doi: 10.1016/j.drudis.2010.11.002. PubMed PMID: 21074634.
CrossRef - Zhao YH, Abraham MH, Le J, Hersey A, Luscombe CN, Beck G, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res.;19(10):1446-57, (2002). Epub 2002/11/12. doi: 10.1023/a:1020444330011. PubMed PMID: 12425461.
CrossRef - Zhao Y, Zheng X, Zhang H, Zhai J, Zhang L, Li C, et al. In vitro inhibition of AKR1Cs by sulphonylureas and the structural basis. Chem Biol Interact.;240:310-5, (2015). Epub 2015/09/13. doi: 10.1016/j.cbi.2015.09.006. PubMed PMID: 26362498.
CrossRef - Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res.;46(W1):W257-W63, (2018). Epub 2018/05/03. doi: 10.1093/nar/gky318. PubMed PMID: 29718510; PubMed Central PMCID: PMCPMC6031011.
CrossRef - Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model; 52(11):3099-105, (2012). Epub 2012/10/25. doi: 10.1021/ci300367a. PubMed PMID: 23092397.
CrossRef - Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res.;42(Database issue):D1083-90, (2014). Epub 2013/11/12. doi: 10.1093/nar/gkt1031. PubMed PMID: 24214965; PubMed Central PMCID: PMCPMC3965067.
CrossRef - Stewart JJ. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J Mol Model.;19(1):1-32,(2013). Epub 2012/11/29. doi: 10.1007/s00894-012-1667-x. PubMed PMID: 23187683; PubMed Central PMCID: PMCPMC3536963.
CrossRef - Ali IO, Salama TM, Bakr MF, El‐Henawy AA, Lateef MA, Guma. Synthesis of nano-sized zeolite-Y functionalized with 5-amino-3-thiomethyl 1H-pyrazole-4-carbonitrile for effective Fe (III)-chelating strategy. HAJRoCI. ;44(9):5193-222, (2018).
CrossRef
Abbreviations
| E: The total energy (Kcal/mol). | I: Ionization potential, |
| LUMO: Lowest Occupied Molecular Orbital (eV) | A; electron affinity; |
| HOMO: Highest Oc cupied Molecular Orbital (eV) | η: Hardness(eV), |
| ΔG: difference between HOMO and LUMO energy levels(eV) | S: Softness(eV), |
| ΔNmax: maximum number of electrons transfer. | χ : Electronegativity (eV); |
| E_score1; Initial free binding energy of the ligand from a given pose. | µ+: electron accepting chemical potentials , |
| ∆E: Final free binding energy of the ligand from a given pose, | µ-: electron donating chemical potentials, |
| E_conf; Free binding energy of the ligand from a given conformer | ω+: electron accepting capacity, |
| E_place. Free binding energy of the ligand from a receptor. | ω-: Electrodonating capacity; |
| E.Int.: Affinity binding energy of ligand with receptor, | ω±: :Electrophilicity index in valance state (eV); |
| H.B.: Hydrogen bonding energy between protein and ligand. | Evdw: Van der Waals energies between the ligand and the receptor. |
| Eele: Electrostatic interaction with the receptor, | RMSD; The root mean square deviation of the pose of the docking pose compared to the co-crystal ligand position. |

