
Nelofar Sediqi1,2 , Aisyah Hasyila Jahidin1*, Mizaton Hazizul Hasan1 and Yuslina Zakaria1
, Aisyah Hasyila Jahidin1*, Mizaton Hazizul Hasan1 and Yuslina Zakaria1
1Faculty of Pharmacy, Universiti Teknologi MARA (UiTM) Cawangan Selangor, Kampus Puncak Alam, Bandar Puncak Alam, Selangor Darul Ehsan, Malaysia, 42300
2Department of Pharmacology, Faculty of Pharmacy, Kabul University, Kabul, Afghanistan, 1006.
Corresponding Author Email: aisya735@uitm.edu.my
DOI : https://dx.doi.org/10.13005/bpj/2250
Abstract
Cancer is one of the most disastrous diseases that leads to a serious threat to millions of people’s health worldwide. Cancer is distinguished by multiple common criteria, known as the “cancer hallmarks" which calcium signaling has either direct or indirect correlation with each of them. An emerging body of evidence suggests that two-pore channels/calcium signaling machinery has a crucial role in the promotion of diverse aspects of cancer, particularly in several cancer hallmarks including cell proliferation, angiogenesis, migration, invasion, metastasis, and metabolic reprogramming. Recent findings linked two-pore channels/calcium signaling machinery with autophagy, chemoresistance, and patients' survival in cancer. The present review provides current findings on the roles of two-pore channels in cancer, particularly in several cancer hallmarks, autophagy, and chemoresistance. Furthermore, a specific focus on recent data concerning the two-pore channels antagonists and novel inhibitors is discussed. This review will furnish readers with a more in-depth understanding of the significance of two-pore channel calcium signalling in cancer and its potential as a druggable target for cancer therapy
Keywords
Apoptosis; Autophagy; Cancer; Cancer Hallmarks; Calcium Signaling; Two-Pore Channels
Download this article as:| Copy the following to cite this article: Sediqi N, Jahidin A. H, Hasan M. H, Zakaria Y. Two-Pore Channels in Cancer Hallmarks: An Update Review. Biomed Pharmacol J 2021;14(3). |
| Copy the following to cite this URL: Sediqi N, Jahidin A. H, Hasan M. H, Zakaria Y. Two-Pore Channels in Cancer Hallmarks: An Update Review. Biomed Pharmacol J 2021;14(3). Available from: https://bit.ly/3l5kTJz |
Introduction
Cancer is a destructive disease resulting in a critical threat to the health of millions of people. This debilitating disease continues to be a major source of morbidity and mortality worldwide in 2020, with 19.3 million new cases and 10 million deaths1. By 2050, the worldwide cancer burden is anticipated to increase to 27 million new cases and 17.1 million deaths2. The malignancy initiates when healthy cells deal with a deficiency in homeostasis and cell regulation, leading to an extensive proliferation of abnormal cells. These abnormal cells further invade the surrounding tissues and eventually spread via circulatory and lymphatic systems to other parts of the human body 2. More than 100 cancer types are identified, each of which is recognized by certain molecular markers, type of genomic alteration, gene-expression profiles, and, respectively, require specific diagnosis and cure. Despite such differences, all cancer cell genotypes share several common pathophysiological characteristics, collectively known as cancer hallmarks. These cancer hallmarks include “self-sufficiency in growth signaling, ability to evade apoptosis, insensitivity to anti-growth signals, capacity to invade and metastasize, limitless replication potential, and promotion of angiogenesis” 3.
Importantly, the direct or indirect role of calcium (Ca2+) signaling in each of the aforementioned processes is well-recognized4,5. Ca2+ signaling is initiated by cytosolic Ca2+ surges as a result of Ca2+ entry from the extracellular space and Ca2+ release from intracellular stores, mostly from the endoplasmic reticulum (ER). The acidic endolysosomal (EL) system involving endosomes and lysosomes also serves as a Ca2+ reservoir. Extracellular ligands trigger Ca2+ release from EL reserves by generating nicotinic acid adenine dinucleotide phosphate (NAADP), a Ca2+-releasing second messenger.In the presence of NAADP, Ca2+ will be released from the acidic stores via two-pore channels (TPCs), a novel superfamily of voltage-gated ion channels6. TPCs have two isotypes in humans, namely TPC1 and TPC2. Both NAADP and TPCs, individually or jointly, regulate a broad number of cellular processes, and besides, their involvement in the pathogenesis of several diseases including cancer has been demonstrated.
The usual strategies for controlling cancer in recent years include surgery, radiation therapy, chemotherapy, immunotherapy, and targeted therapy2,7. The application of these treatment modalities in clinical settings is limited by diverse challenges including adverse drug reactions, potential toxicity, cost-related problems, and resistance. Hence, there is an immediate requirement for a novel, safer, and efficient antitumor agents for the treatment of cancers2. Noteworthy, about 600 cancer drivers have been identified, but a large number thereof remains to be targeted by antitumor agents8.
An increasing body of research has recently discovered a potential link between TPCs and cancer. However, deep investigations for characterizing the connection between TPCs and cancer-relevant processes and mechanisms underlying it in different cancer cells are still in its early stages. In this review, we look at the current role of TPCs in cancer, concentrating on the correlation between TPCs and several hallmarks of cancer, and TPCs’ connection with autophagy in cancer. Additionally, we also provide a brief review of novel TPCs inhibitors and their role in overcoming chemoresistance.
Cancer in the light of Ca+2signaling
According to the World Health Organization (WHO) definition, “Cancer is a common name for a wide class of diseases described by the rapid and uncontrolled formation of abnormal cells that grow beyond their normal barriers, and which can then invade surrounding cells and neighboring tissues, and spread to other organs.”9.Originally, excessive cell proliferation and deficiency in cell death mechanisms together play a central role in malignancy. Essentially, they abolish the homeostasis of tissue and cell growth and eventually lead to cancer development 10.
Intracellular Ca2+ ion is the most abundant and unique ion, which has a huge concentration gradient across the plasma membrane of basically all cells. Intracellular Ca2+ homeostasis is an energy-dependent incident and required a regulated Ca2+signaling motor 11. Ca2+ signaling is the participation of Ca2+ ion in signal transmission for cellular communication and the performance of the cellular physiological processes 12. Thus, Ca2+signaling is involved in the broad pathological conditions in metabolism, neuron degeneration, immunity, and malignancy 13.
Major studies uncovered the implication of Ca2+signaling in the whole process of tumor progressions including proliferation, apoptosis, tumor growth, metastasis, invasion, angiogenesis, and resistance to anti-growth signals 14. Dysregulation of Ca2+signaling in cancer cells alters their physiology and specified them from non-malignant cells15. Similarly, Ca2+ homeostasis is also vulnerable to derailment by several oncogenes and tumor suppressors. Tumors remodel their Ca+2 signaling network to enable them to grow erratically, metastasize, evade apoptosis, survive immune-attack, or generate neovascularization 16,17. A plethora of research groups worldwide supported the alterations of Ca2+signaling and Ca2+ channels and pumps in cancer cells, as well as the intersection of Ca2+signaling particular processes with various cancer hallmarks. Additionally, studies highlighting novel pharmacological modulators of Ca2+ pumps and channels suggest that Ca2+ machinery is amenable to targeting by pharmacological agents. These studies implement insight into targeting Ca2+ channels and pumps as a potential therapeutic intervention against cancer18.
The EL Ca2+ store and Ca2+ signaling
The EL system which is part of a growing family of acidic calcium stores involves lysosomes, endosomes, and autophagosomes19. Lysosomes provide a center for macromolecular degradation and recycling, endocytic recycling, and the regulatory process of cellular nutrients. Being the second-biggest Ca2+ store, lysosomes also serve as a crucial Ca2+ signaling system for the cell with a concentration of free Ca2+ in the range of 500 to 600 µM in various mammalian cells, approximately 5000-fold higher than the cytosolic Ca2+ content 20.
Endosomes are dynamic, specialized compartments that primarily function to sort and traffic cargoes internalized into the cells21. They also serve as key signaling organelles that release Ca2+ to initiate signaling cascades. Besides, they require Ca2+ at several steps of their maturation, including lysosomal fusion, the fusion between late endosomes and lysosomes, and the reconstruction of lysosomes from hybrid late-endosomal–lysosomal organelles22.
Acidic EL Ca2+ homeostasis is regulated by concerted actions of various channels and pumps. EL Ca2+ content is maintained by an unknown Ca2+/H+ exchanger or Ca2+ transporter. While TPCs and transient receptor potential mucolipin 1 (TRPML1) are the primary Ca2+ channels responsible for releasing Ca2+ from the EL stores 6,20. Ca2+ released via EL Ca2+ channels leads to small, localised Ca2+ signals. Ca2+-induced-Ca2+-release (CICR), a process that triggers the release of Ca2+ from the ER, can amplify these signals into larger, global signals 23.
TPCs—EL Ca2+ release channels
TPCs form a family of ligand and voltage-gated ion channels24, which belong to the group of endolysosomal membrane proteins. Their names are derived from the two-pore domains that are found on each subunit. TPC subunits consist of 12 transmembrane domains (TMDs) with the putative pore loops between TMD5/6 and TMD11/1225. Structurally, each dimer of tandem Shaker-like domains dimerize to form a functional channel26. TPCs have three isoforms, but only two, designated TPC1 and TPC2 are present in humans. TPC1 essentially locates in the endosome while TPC2 resides in the lysosome27, and they are encoded by TPCN1 and TPCN2 genes, respectively28.
It is well known that TPCs mobilize Ca2+ out of EL stores upon stimulation by NAADP29. However, in 2012 and 2013, two independent investigations disproved the long-accepted claim by other researchers30,31. Both publications argued that TPCs are (sodium) Na+ release channels that are stimulated by phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) and inhibited by mammalian target of rapamycin (mTOR). Other than Ca2+ and Na+, TPCs have also been shown to be permeable to hydrogen (H+) and potassium (K+)32,33. In addition, various stimulators of TPCs other than NAADP and PI(3,5)P2 have been identified, including leucine-rich repeat kinase 2 (LRRK2) and action potentials34,35. Meanwhile, inhibitors of TPCsinclude Ca2+ and Na+ ion channel blockers, as well as magnesium ion (Mg2+)36. Particularly, Mg2+ inhibited the PI(3,5)P2-mediated TPCs activation in a concentration-dependent manner 37.
Functional roles of TPCs
TPCs perform vital functions at the cellular and organism levels. At the cellular level, they organize cellular transportation, pH regulation, and cell membrane stimulations. At the organism level, they are linked with different physiological and pathological processes, such as hair pigmentation, Ebola viral infection, and cancer growth38. Particularly, they are associated with multiple physiological processes, for instance, cytokinesis, fertilization, embryogenesis, cell differentiation, and autophagy36. Recent investigations have demonstrated the involvement of TPCs in tumorigenesis6,8,39-41. Both isoforms of TPCs were found expressed in various cancer cell lines isolated from blood, bladder, breast and, liver cancers. Furthermore, in the SKBR3 human breast cancer cell line, the expression of TPC1 was almost 3 – 7 folds more than the expression of TPC242. Apart from that, NAADP-mediated Ca2+ release via TPCs has also been shown to be crucial for immune cell function43. TPCs also play a role in endolysosomal activities and receptor-trafficking44-46. In the absence of TPC1, uptake of toxins in early endosomes is diminished. Another study communicated the significance of TPC1 and TPC2 during starvation30. Through direct interaction with mTOR, TPCs can sense and regulate cellular nutrient status. The ability to endure physical challenges during starvation is greatly compromised in the absence of both TPCs and mTOR. Additionally, TPC2 has also been shown to control melanin synthesis by regulating the pH and size of melanosomes 47.
Implications in diseases
Numerous studies established TPCs association with some pathological conditions, which has been reviewed elsewhere48.Evidence from models of Parkinson’s disease, cardiac dysfunction, cancer, non-alcoholic fatty liver disease, diabetes, and Ebola infection showed that TPCs are linked to these disorders 49.The association of lysosomal Ca2+signaling to several degenerative diseases such as Parkinson’s disease and Alzheimer’s disease was later confirmed to be related to TPC220.Furthermore, TPCs are connected with metabolic disorders related to the deficiency of endolysosomal trafficking. TPC2 knockout (KO) mice are highly susceptible to hepatic cholesterol overload and liver damage consistent with non-alcoholic fatty liver hepatitis, likely due to abnormal hepatic cholesterol handling50. Various studies have also evinced the role of TPCs as the main element in the pathogenesis of several viral infections, including Ebola virus (EBOV), Middle East Respiratory Syndrome-Coronavirus (MERS-CoV), and Merkel cell polyomavirus (MCPyV) infection28,51,52. Blocking TPCs, particularly TPC2 using tetrandrine can inhibit SARS-CoV-2 replication in host cells53,54. This shows that TPC2 could have a role in preventing the spread of coronavirus disease2019 (COVID-19).
TPCs in cancer cells
The distribution of TPCs has been characterized in various normal and cancer cells. Brailoiu and colleagues42 have shown that both TPC1 and TPC2 are expressed in SKBR3 and PC12 cells. The quantitative PCR (qPCR) has demonstrated that between the two TPCs isoforms, the TPC1 isoform was the main expressed isoform in PC12 and SKBR3 cells. Particularly, TPC1 transcription was roughly three to eight times higher than TPC2 transcription in SKBR3 cells.This claim is supported by another independent study showing that the expression of TPC1 mRNA is approximately 50 folds more than TPC2 in primary cultures of human metastatic colorectal cancer (mCRC)6. In contrast, a study by Jahidin et al.55 has described similar expressions of TPC1 and TPC2 transcript in various sets of human breast cancer cells, including the MCF-7, T47D, ZR-75-1, BT-483, SKBR3, MDA-MB-231, and MDA-MB- 468 cancer cells. The results also showed that TPC1 and TPC2 were expressed similarly in the abovementioned tumorigenic cell lines and non-tumorigenic cell lines, namely 184B5 and 184A1. A subsequent study by Nguyen et al.41. Compared the expression of mRNA level of TPCs in different cell lines against the MDA-MB-231 human breast cancer cells, namely T24 (bladder cancer), Jurkat (leukemia), and Huh7 (liver cancer) cells. The authors exhibited that TPC2 mRNA is expressed in all the three studied cell lines. Meanwhile, the mRNA level of TPC1 is highly expressed in T24 and Huh7 but Jurkat cells. Moreover, the authors also displayed the functionality of TPCs in T24 cells. Using endolysosomal patch-clamp experiments, Nguyen and colleagues demonstrated that tetrandrine, a pharmacological inhibitor of TPCs, drastically reduced PI(3,5)P2-elicited currents.Another study revealed a higher expression of TPC2 mRNA in a drug-resistant leukemic cell line56. The presence of 2-fold TPC2 in vincristine-resistant CEM cells (VCR-R CEM) is important for cell proliferation. The KO of TPC2 in these cells resulted in a significantly slower growth compared to the wild type (wt). Moreover, heightened sensitivity to vincristine was observed in these TPC2-deficient cells. TPC2 was also overexpressed in various oral squamous cell carcinoma cell lines 57and human skin cutaneous melanoma (SKCM)39. Interestingly, TPC2 expression in SKCM is inversely correlated to metastasis39. It was found that the expression of TPCN2 mRNA in metastatic patients was significantly lower than in the primary patients. Additionally, elevated levels of mesenchymal markers such as vimentin and ZEB-1 were observed in TPC2 KO cells, suggesting distinct roles of TPC2 in primary and metastatic tumors of melanoma. Collectively, these data suggested that expressions of TPC1 and TPC2 are cell- and stage-specific and are associated with cancer development and metastasis.
TPCs and cancer hallmarks
The significantroles and close relationship between TPCs and cancer hallmarks particularly, proliferation, migration, invasion, angiogenesis, and metastasis have been reviewed of late 19,58-62. According to several studies, NAADP/TPC/Ca2+ signalling has been implicated in a variety of cancer-related processes, ranging from carcinogenesis to metastasis6,19,39-41.TPCs play a crucial role in cancer progression, and the inhibition of TPCs activity via pharmacological agents or gene-silencing techniques leads to the elimination of cancer-related processes such as angiogenesis40,61,63. Furthermore, the loss or reduction in TPC activity is associated with the elimination of cancer cell migration41 and neoangiogenesis64. The following sections will dive into more details on the roles of TPCs in each of the hallmarks of cancer.
TPCs and cancer cells proliferation
Sustained proliferation is the main feature of cancer cells. There is a highly-regulated system in healthy cells that controls the normal cell cycle process resulting in accurate cell growth and function. In contrast, malignant cells mislead the cell cycle by mutations of some genes including the TP53 and retinoblastoma (Rb) that head towards an extreme proliferation ability 57. As Ca2+ is a multifunctional second messenger to both proliferation and cell death, any defect in Ca2+signaling or Ca2+ concentration regulation may cause uncontrolled proliferation and inhibition of apoptosis and consequently, contributes to tumorigenesis65.
The correlation between the NAADP/TPC2/Ca2+ system with cancer hallmarks including the proliferation, invasion, metastasis, and angiogenesis was studied in in vitro and in vivo models specifically, in xenografted murine models with B16 melanoma cells40. The data showed that NAADP-mediated Ca2+signaling is critically important for neoangiogenesis, formation of metastasis, and tumor growth. Using Ned-19, the pharmacological antagonist of NAADP on melanoma cells and in mice inoculated with B16 cells altered metastatic behavior and extremely diminished development of lung metastasis. These probably due to the impeded entry of melanoma cells into the blood circulation as a result of reduced vascularization of the primary tumor by Ned-19. Moreover, the Ned-19 treatment significantly upregulated E-cadherin and downregulated N-cadherin. These two founding members of the cadherin superfamily are key modulators of tumorigenesis66. Cadherin-switching from E-cadherin to N-cadherin signals for invasion, migration, and metastasis67. However, this process can be reversed with the re-establishment of E-cadherin and hence the acquisition of epithelial phenotype.
Cytokinesis is the final and most tightly controlled phase in cell division, therefore, any dysregulation in this process can result in multinucleation and aneuploidy, processes that can lead to chromosomal instability and tumorigenesis. According to Horton et al.68, TPC1-overexpressing cells compared to wt, displayed cytokinetic abnormalities, multinucleation, and enlargement the cytokinetic defects, multinucleation, and enlargement, and additionally, cancer tissues expressed considerably more TPC1 than normal tissues.
TPC2 has been implicated in the modulation of skin pigmentation in Xenopus oocytes in several studies 69,70. The authors demonstrated that overexpression of TPC2 induced pigmentationdefects. Besides, the studies have identified that polymorphisms in the TPC2N gene caused some genetic variants. Collectively, these studies suggested that TPC2 overexpression lowered melanin production and enhanced skin cancer susceptibility.
The role of NAADP/TPCs-Ca2+signaling in mCRC cells derived from a human liver metastasis has recently been explored6. The researchers have found that EL Ca2+ storemediates calcium release by activation of the PI3K/AKT and ERK signaling pathways in mCRC cells (Figure 1). The inhibition of TPCs, in particular, the TPC1 isoform, which is the more abundant isoform of TPCs in mCRC cells, has completely decreased Ca2+ release and proliferation of mCRC cells.
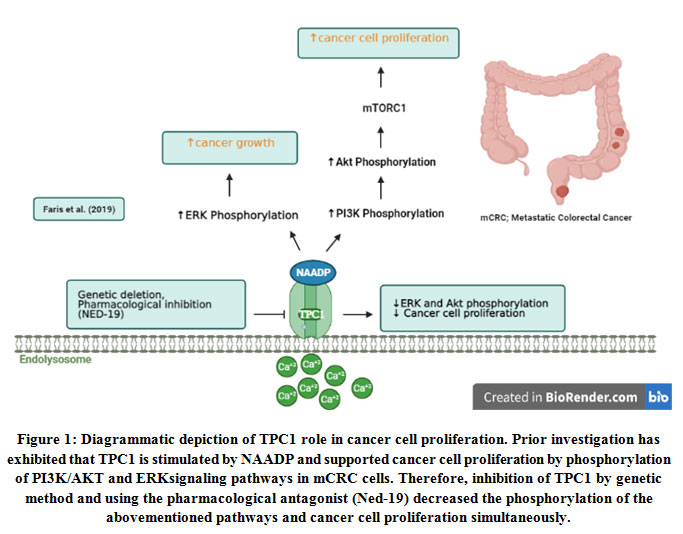 |
Figure 1: Diagrammatic depiction of TPC1 role in cancer cell proliferation. Prior investigation has exhibited that TPC1 is stimulated by NAADP and supported cancer cell proliferation by phosphorylation of PI3K/AKT and ERK signaling pathways in mCRC cells. |
Similarly, Sun et al.71assessed the role of TPC2 in two cancer cell lines, 4T1 mouse breast cancer cells, and HeLa human cervical cancer cells. Interestingly, they found that proliferation is reduced in TPC2 knockdown cells compared to control or TPC2 overexpressing cells. More recently, Müller et al.8 have identified TPC2 as a cancer driver. The findings have shown that genetic deletion of the channel reduced the proliferation of liver cancer cells in vitro and successfully stopped tumour growth in an ectopic mice model.
TPCs and cancer cell angiogenesis
Angiogenesis is an important process in cancer development. It is the most important phase in the progression of a benign tumor to malignancy. Generally, vascular endothelial growth factor receptor (VEGF) and its receptor, vascular endothelial growth factor receptor 2 (VEGFR2), are two key molecules for initiating and promoting angiogenesis. They play a central role in activating the angiogenesis process, including the vascularization of tumors40. During angiogenesis, tumor cells release VEGF that stimulates signal transduction and Ca2+signaling, and finally, leads to endothelial cell proliferation19,64.
According to Favia et al.64, there is an association between NAADP/TPC2/Ca2+signaling pathway with VEGF-induced neoangiogenesis. The VEGFR2/NAADP/TPC2/Ca2+signaling pathway is crucial for VEGF-induced angiogenesis, according to results from in vitro and in vivo studies.The findings of the in vitro investigation showed that human umbilical vein endothelial cells (HUVEC) transfected with TPCN2 shRNA failed to construct a tubular network. A similar outcome is observed when NED-19, the pharmacological antagonist of TPCs is applied. Likewise, this process is also halted in vivo. The results of the matrigel plug assay in mice showed that Ned-19 had an inhibitory effect on the construction of the vessels. TPC2 KO animals replicate this observation, whereas TPC1 KO mice do not.Interestingly, TPCN1-transfected-mice induced excessive vessel construction of plugs in 5 days.
VEGF-induced vessel formationis also impaired in the presence of naringenin, a flavonoid known to inhibit both TPC1 and TPC263. To establish the role of naringenin and TPC signalling on angiogenesis, the author used matrigel matrix and C57BL/6 mice in both in vitroand in vivo investigations.The results confirmed that the anti-angiogenic effect of naringenin occurred by affecting the VEGF-NAADP/TPC2- Ca2+signaling.
VEGF stimulates endothelial-relevant processes including the proliferation of endothelial cells, survival, migration, and vascular homeostasis through the ERK1/2 phosphorylation cascade maintained through endogenous Ca2+ release via TPC2 and InsP3R and as well as through extracellular Ca2+ entry via TRPC4, NCX1, and TRPC3, and the SOCE pathway. Hence, the pharmacological blockade of TPC by using the NED-19 is a profitable way to decelerate tumor vascularization 61,64.
TPCs and migration, invasion, and metastasis
Approximately 90% of cancer-related mortalities correlate to metastatic cancer72.Metastasis is summarized as a two-phase process that includes the translocation of cancer cells to the dissemination site, and the ability to colonize secondary sites and establish metastatic lesions by primary tumor cells. The main steps in tumor metastasis are the loss of cell-cell connections, the transition of original cancer cells into migratory mesenchymal cells, as well as cancer cells invasion. The disturbed environment due to immune attacks, lack of oxygen, blood supply and nutrients, accumulation of lactic acid, and increased cell death are among the reasons why cancer cells metastasize for survival73. Ca2+signalinghas been linked to a number of basic pathophysiological events related to cancer metastasis and progression19. Intracellular Ca2+ channels including IP3Rs, TRPMLs and TPCs, have been associated with these processes6,19,59,74.
An in vitro result demonstrated that TPC1 and TPC2 silencing diminished the adhesion and migration of T24 human urinary bladder cancer cells41. Similar observations were noticed when pharmacological inhibitors of TPCs were applied. Treatment with Ned-19 abrogated migration of T24 and Huh7 cells. Likewise, the migration of T24, Huh7, and 4T1 mouse breast tumor cells was also impeded in the presence of tetrandrine. Further analysis in a mouse model inoculated with 4T1-Luc cells revealed reduced formation of lung metastasis upon treatment with tetrandrine or Ned-19, and as well as TPC2 silencing.The authors suggested that these observations were associated with the disruption of β1-integrin trafficking in the EL system. Ample trafficking of β1-integrin is critical for initiating promigratory mechanisms, thus hindering the construction of leading edges required for the migration of invasive cancer cells. Additionally, Ned-19 treatment controls the migratory and adhesive abilities of B16 cells40,61. Besides, the pretreatment of B16 cells with Ned-19 diminished the expression of N-cadherin and E-cadherin, thus modulating the cell migratory behavior and invasion ability of B16 cells40.
The attenuation of TPC transcript in metastatic patients compared to primary patients indicated a distinct predictive role of TPC2 in these two stages of cancer39. In contrast to the abovementioned studies, D’amore et al.39 demonstrated that TPC2 KO in CHL1 cells, a model of human amelanotic melanoma cells derived from a metastatic site increases the cells ability to migrate and metastasize (Figure 2). The authors also exhibited that TPC2 KO enhanced the secretion of matrix metalloproteinase 9 (MMP9) by CHL1 cells compared to the wt. TPC2 KO cells also upregulated the expression of mesenchymal markers, namely zinc-finger E-box-binding homeobox1 (ZEB-1), vimentin and N-cadherin. Additionally, TPC2 KO cells also expressed a higher level of Melanocyte Inducing Transcription Factor (MITF), which promoted melanoma cell survival and migration. D’amore and colleagues39 have also studied the correlation of TPC2 and Yes-associated protein 1 (YAP)/ Transcriptional coactivator with PDZ-binding motif (TAZ) pathway in human SKCM. The YAP and TAZ are transcriptional coactivators that regulate several processes such as cell proliferation, apoptosis, angiogenesis, tumorigenesis, and metastasis75. The activation of these transcriptional coactivators is correlated with enhanced metastatic capability76. D’amore and colleagues39 revealed that TPC2 KO cells upregulated the target genes of YAP/TAZ, namely ankyrin repeat domain-containing protein (ANKRD1), cysteine-rich 61 (CYR61), and connective tissue growth factor (CTGF). Moreover, translocation of YAP/TAZ from the cytoplasm to the nucleus was observed in TPC2 KO cells. In addition, the expression of ORAI1 and PKC-βII are downregulated in TPC2 KO cells. Both ORAI1 and PKC-βII negatively regulate YAP/TAZ activity. Collectively, these results indicated that TPC2 KO activates YAP/TAZ, thus contributes to the aggressiveness and invasiveness of metastatic melanoma.
 |
Figure 2: Diagrammatic depiction of TPC2 role in metastatic melanoma. The previous finding showed that TPC2 expression is reduced in the late stage of melanoma and is associated with increased aggressiveness of melanoma cells. |
TPCs and metabolic reprogramming in cancer
Metabolic reprogramming is one of the cancer hallmarks.During cancer progression, metabolic pathways are altered in response to intrinsic and extrinsic cues like mutated enzymes and hypoxia, respectively77. This is crucial to enable tumor cells to escape immune system supervision, survive and proliferate uncontrollably78.Cancer cells reprogram their catabolic and anabolic metabolism pathways to initiate and develop malignancy79. It has been shown that dysregulation of Ca2+signaling results in the alteration of metabolic pathways in cancer cells that simultaneously supports aggressiveness. For instance, mitochondrial calcium uniporter (MCU) expression is associated with mitochondrial Ca2+ uptake in promoting hypoxia-inducible factor 1-alpha (HIF-1α) expression and invasiveness and, consequently, promote tumor size and metastasis in triple-negative breast cancer80. It has been exhibited that TPC2 is associated with metabolic reprogramming in cancer cells81. The findings have shown that TPC2 overcome glucose utilization for energy production in liver cancer cells. As depicted in Figure 3, the mechanism that TPC2 uses for this purpose is the regulation of the main mediator of glycolytic flux, namely hexokinase II (HK II). TPC2 KO results in diminished phosphorylation of HK II. Healthy cells convert glucose to pyruvate in the cytosol by glycolysis, then further oxidize it to carbon dioxide in mitochondria, while paradoxically, various cancer cells use aerobic glycolysis or the Warburg effect. At the time of TPC2 inhibition through pharmacological agents or gene KO of this channel, the examinations of extracellular flux have shown a shift towards lower glycolysis and, thus, liver cancer cells metabolic reprogramming was partially reversed8.
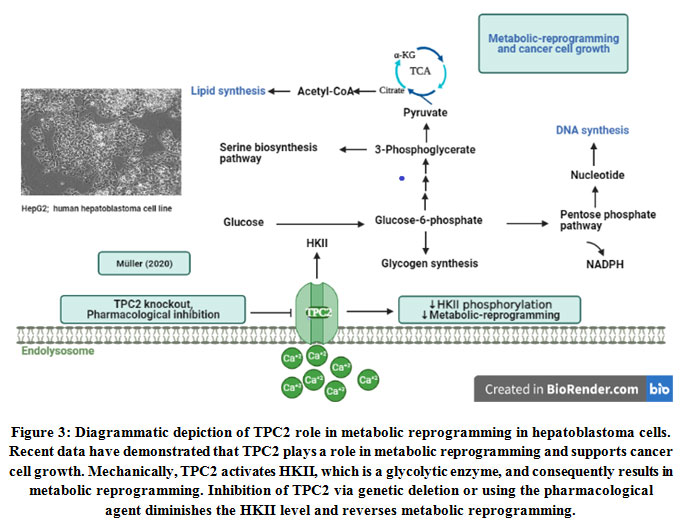 |
Figure 3: Diagrammatic depiction of TPC2 role in metabolic reprogramming in hepatoblastoma cells. Recent data have demonstrated that TPC2 plays a role in metabolic reprogramming and supports cancer cell growth. Mechanically, TPC2 activates HKII, which is a glycolytic enzyme, and consequently results in metabolic reprogramming. |
TPCs and autophagy in cancer
Autophagy is an essential cellular catabolism and recycling process that aims to maintain homeostasis where eukaryotic cells sequester unnecessary intracellular components, including damaged molecules, damaged organelles, and foreign materials, transported to lysosomes in vesicles for degradation82,83. A plethora of evidence indicates the bipolar nature of autophagy in cancer. In the benign and tumor initiation stages, autophagy demonstrated a preventive role as a tumor-suppressive mechanism. As cancer progressed, this role changes from suppressing to promoting tumor development. Cancer cells expanded autophagy dependency as metabolic and biosynthetic requirement increases simultaneously82. A majority of studies have unveiled the link between autophagy and the hallmarks of cancer, including sustaining proliferation, epithelial-mesenchymal transition, tissue invasion, and metastasis84-93. Additionally, several investigations have reported the involvement of autophagy in reprogramming Tumor metabolism, as well as resistance to therapy94-111. The investigations have shown that both basal and induced autophagy is regulated by Ca2+signaling19.
Along with other calcium-permeable channels that have been implicated in the autophagy regulation of various cells, TPCs are also involved9,112. It has been reported that TPCs regulate autophagy either positively or negatively71. The distinct performance of TPC is likely related to factors like cell type, protein expression, and cell condition. Lin et al113demonstrated that TPC2 modulates the autophagy process in mouse skeletal muscle, in contrast to a previous study by Cang et al.30 which suggested that both TPC1 and TPC2 play no role in autophagy. This discrepancy could be due to various factors, including strategies used to develop the TPC KO mouse model, as well as the diets and husbandry of the animal113. In the study by Lin and colleagues113, increased autophagy-flux and agglomeration of microtubule-associated protein light chain 3 (LC3) proteins were observed. Besides, the authors also reported that loss of TPC2 had caused an abnormality in lysosomal pH and acid enzymatic activity, which are important for the autophagy process. In agreement with Lu et al.114, this study showed that overexpression of TPC2 had blocked the autophagosomal-lysosomal fusion in HeLa cells or mouse embryonic stem cells, resulting in the accumulation of autophagosomes. This is corroborated by the observation that cells lacking TPC2 either by knockdown technique or treatment with TPCs antagonist Ned-19 exhibited a low accumulation of autophagosomes. In line with this, researchers have detected that TPC2/NAADP/Ca2+signaling inhibits autophagy through lysosomal alkalinization 50,114. In the 4T1 mouse breast and HeLa human cervical cancer cell lines, TPC2 overexpression has reduced the autophagosome-lysosome fusion and consequently led to the accumulation of LC3-II and syntaxin 17 (STX17)-positive autophagosomes19,71. By contrast, Pereira et al.115 have found that TPC downregulation has inhibited the enhancement of glutamate-induced autophagic flux, a central nervous system excitatory neurotransmitter. They have investigated the involvement of NAADP/TPCs calcium signaling machinery in glutamate-induced autophagy in rat astrocytes and SHSY5Y neuroblastoma cells. Remarkably, TPC inhibition by gene-silencing and application of NED-19 have limited the glutamate-induced increase in autophagic flux in those cells. Hence, when the cells are treated with lysosomal inhibitors, the LC3-II levels in TPC-downregulated cells and cells with pretreatment of NED-19 are reduced or failed to increase.
TPCs and the cancer patients’ survival
Individualized therapy is an effective strategy for the management of cancer particularly, aggressive cancers. Therefore, finding the predictive biomarkers is an important step to improve the selection for patients going for a precise treatment such as chemotherapy, targeted-agents, or immunotherapy according to their benefits. Previous studies have shown the correlation between calcium channel gene-expression with cancer patients’ survival. For example, a high gene expression of three genes of L- and T-type calcium (CACNA1D, CACNA1F, and CACNA1H) was associated with poor survival of ovarian cancer patients 116. There is evidence that showed the direct or indirect contribution of TPCs to cancer-patients survival. The amplification of 11q13 occurs in a variety of malignancies, including half of all oral squamous cell carcinomas (OSCC), resulting in a poor outcome. Huang et al.57 have found overexpression of multiple genes, including TPCN2, which contributed to the amplification of 11q13 and related to the poor outcome of cancer patients. D’Amore et al.39 have shown an inverse correlation of YAP/TAZ activity with TPC2 expression in SKCM. YAP and TAZ are transcriptional coactivators that regulate tumorigenesis processes in various cancers117. Interestingly, YAP hyperactivity is related to a weakcancer prognosis. Hence, by reducing or inhibiting TPC2 expression, YAP/TAZ activity is increased and consequently, cancer outcome is decreased39. In contrast, Shivakumar et al.118 have described a direct association between TPCN2 expression and bladder cancer survival. The study discovered an epigenetic connection between DNA methylation and microRNA, which was linked to gene expression and might be used as a prognostic marker in bladder cancer. They found that inhibition of TPCN2 expression is highly associated with a better survival outcome19,49,118. According to Li et al.119 and Muller8,a set of novel differentially expressed gene that predicts biochemical recurrence after prostatectomy has demonstrated a higher gene expression of TPC2, which is related to a poor survival probability of prostatic adenocarcinoma patients.
Targeting TPCs in cancer
Owing to the importance of TPCs in physiological processes such as cell growth and pathophysiological conditions such as cancer progression, previous studies strongly recommend that TPC1 and TPC2 display suitable molecular targets. Hence, various pharmacological modulators of TPCs targeting the cancer cells are deeply reviewed19,48,120.
NED-19
Ned-19 is a synthetic inhibitor of TPCs19, recognized for its importance in various conditions, including tumorigenesis, metastasis, Ebola infection, autophagy of hepatocytes during liver injury, and VEGF-induced angiogenesis48. It is a membrane-permeant noncompetitive NAADP antagonist that acts selectively but indirectly similar to NAADP to exert its inhibitory effect on TPC48. Originally, Naylor et al.121 had recognized Ned-19 through ligand-based virtual screening targeted against NAADP. A majority of studies have demonstrated the antagonistic effect of Ned-19 against TPCs via its effect on different hallmarks of cancer in various cancer models. For instance, the application of Ned-19 (250, 150 µmol/L) for 8 and 16 h significantly diminished the migration of T24 and Huh7 cells41. Ned-19 has also been shown to impair the invasion of T24 cells [41]. In an in vivo mice model of breast cancer cells, Ned-19 (150 µmol/L for 24 hrs) has lessened the formation of lung metastasis19. The in vitro and in vivo anti-angiogenic impact of Ned-19 (100 µmol/L for 30 min, 25, 50, 100 µmol/L for 4 weeks) was observed in HUVECs and murine models 64. In an in vitro model of mCRC cells, Ned-19 (100µ mol/L for 30 min) has blocked TPC1 and consequently, reduced cancer cell proliferation6. Furthermore, the treatment of Ned-19 (5 mg/kg for 4 weeks) has abolished the NAADP-induced Ca2+ release and decreased the tumor growth and vascularization of melanoma40,61. Besides, the pretreatment of B16 cells with Ned-19 (25, 50, and 100 μmol/L for 30 min) has reduced N-cadherin and increased E-cadherin expression, modulating the cell migratory behavior and invasion ability of B16 cells. Moreover, the treatment of B16 cells with Ned-19 (25, 50, and 100 μmol/L for 5 hr) has inhibited viability, proliferation, and expression of VEGFR2 in B16 melanoma cells 40.
Tetrandrine
Tetrandrine is a bisbenzytehahydroisoquinolinealkaloid originally derived from Stephania tetrandra. Both extraction and chemical synthesis are the main sources of tetrandrine8. This compound is reported to have numerous medicinal properties including anti-inflammatory122, antinociceptive123, antifibrotic124, antidepressant125, and antiadipogenic effects126. Pharmacologically, tetrandrine is classified as L-type Ca2+ channel blockers. It is one of the L-type Ca2+ channel blockers tested for activity against EBOV, apart from diltiazem, nimodipine, and verapamil. Compared to the aforementioned pharmacological agents, tetrandrine has exhibited the highest potency (IC50 = 55 nM)48. Additionally, tetrandrine has also been reported to effectively prevented other infections directly acting on TPCs, including the MERS-CoV and SARS-CoV-2 viruses8. A plethora of studies have shown the inhibitory effect of tetrandrine on cancer hallmarks in different cell lines via TPCs blockade. For instance, the treatment with tetrandrine effectively inhibited the proliferation of VCR-R CEM cells (IC50: 5 – 15 µM, 48 h)56. Besides, it abolished the migration of cancer cells, including the T24 (15 mmol/L for 8 h), Huh7 (2.5 mmol/L for 8 h), and 4T1 (10 mmol/L for 8 h) cells. Furthermore, tetrandrine (15 mmol/L for 8 h) has impaired adhesion in T24 cells41.Moreover, in vivo administration of tetrandrine (10 mmol/L for 24 h) employing a mouse model of mammary cancer cells has reduced the formation of lung metastases19,41. Furthermore, tetrandrine can abolish the metastatic ability of murine cancer cells in vitro and in vivo41,48. A more recent finding has shown the superiority of tetrandrine congeners over other known TPC2 inhibitors such asNed-19, and naringenin in inhibitingcancer hallmarks, including the impairing of proliferation and proangiogenic signaling. The pharmacological efficacy of tetrandrine in various disease models, however, is limited by several shortcomings, including partly moderate inhibition of TPC2 (54% at 10 µM), the complexity of its structure, and its toxicities (hepaticand pulmonary toxicities)8. Additionally, it has multiple targets that are limited in animal models and not yet applicable for clinical settings worldwide120.
Naringenin
Naringenin (5,7-dihydroxy-2-(4-hydroxyphenyl)chroman-4-one) is a naturally occurring flavonoid found primarily in fruits like grapes and oranges. This compound has been shown to possess various therapeutic properties, including antioxidant, anti-inflammatory, chemopreventive, and antidegenerative127, as well as antiangiogenic128.Pre-clinical investigations have demonstrated that naringenin and its precursor naringin have the potential to treat a wide range of metabolic and cardiovascular disorders129. The chemopreventive and antitumor effects of naringenin have also been displayed in multiple experimental models of cancers, including breast130, oral131, and colon132. Interestingly, naringenin has been proposed to serve as a potential pharmacological weapon against coronavirus infection133,134.Recently, naringenin has been introduced as a novel inhibitor of TPC1 and TPC263.The authors used HUVEC cells to demonstrate that naringenin inhibits VEGF-dependent Ca2+signaling. The cells were pretreatedwith varying doses of naringenin followed by stimulation with 100 ng/mL VEGF to induce Ca2+ release. Naringenin with an IC50 of roughly 200 μM, considerably attenuated Ca2+ mobilization in a dose-dependent and reversible manner.
Further analysis using Ca2+ imaging experiments showed that naringenin did not significantly impair the phosphorylation of VEGFR2, which confirmed that the inhibition occurs downstream of the receptor. Additionally, naringenin’s action is also limited to NAADP-mediated Ca2+ release as shown by disruption of histamine-evoked Ca2+ release which is NAAADP/TPC2-dependent. Similarly, a significant reduction of intracellular Ca2+ release induced by NAADP-AM, the cell-permeable form of NAADP, was also observed with 500 and 1000 uM of naringenin. Whilst no effect was detected on ATP-evoked Ca2+ release, which is IP3-dependent and NAADP-independent. The selectivity of naringenin on the NAADP/TPC2 pathway was further corroborated by the application of Ang-1, an angiogenic agonist known to stimulate Ca2+ release independent of NAADP. The results showed that the mobilization of Ca2+ by Ang-1 was not altered by either 500 or 1000 uM of naringenin.
Novel antagonists
Due to the toxicity of tetrandrine in animals, Müller et al.56 have synthesized a library of bisbenzylisoquinoline derivatives (BBIQDs) to screen for novel antagonists of TPCs with less toxicity. Two small molecules hit were identified from this screening, namely SG-005 and SG-094. Using the whole endolysosomal patch-clamp method, the authors tested the efficacy of these two molecules in inhibiting TPC2 function. SG-005 showed similar potency with tetrandrine with 50% reduction of TPC2 density. Meanwhile, SG-094 exhibited higher potency with 70% reduction. These novel inhibitors have some advantages over tetrandrine, including the simplification in the synthesis, similar or improved inhibition (SG-094: 75% at 10 µM; SG-005: 44% at 10 µM) of PI (3,5)-P2-evoked TPC2 currents and, comparable or more potent antiproliferative activities against multiple cancer cell lines8. An extreme antiproliferative effect on RIL175 cells was observed with administration of SG-005 (IC50: 2.4 µM) and SG-094 (IC50: 3.7 µM), compared to tetrandrine (IC50: 9.1µM)8. The authors also showed that both SG-005 and SG-094 considerably outdid the performance of Ned-19 and naringenin, which failed to exhibit antiproliferative activities even at a concentration of more than 75 µM. Another in vitro study using VCR-R CEM demonstrated the efficacy (IC50: 5 – 15 µM, 48 h) of SG-005 and SG-094 in impeding the proliferation of this cell line56. Müller8 also reported that a far lower dose of SG-005 and SG-094 (10 µM each) was enough to prevent the phosphorylation of VEGFR2 downstream targets when tetrandrine showed no significant effect. Additionally, a higher dose of NED-19 (100 µM) and naringenin (500 µM) were required to prevent VEGF-induced vessel formation. The toxicity of both SG-005 and SG-094 was also evaluated in vitro and in vivo. Müller et al.56 performed propidium iodide exclusion assays on peripheral blood mononuclear cells (PBMCs, n = 3 healthy donors) and demonstrated reduced toxicity of SG-094 (< 5% dead cells) compared to tetrandrine and SG-005 (25% dead cells). Moreover, SG-094 was also shown to be well tolerated when applied to a mouse model (90 nmol/kg/d) for three consecutive days.
Another two novel inhibitors of TPC2 recently identified were flavonoids MT-8 and UM-9 135. MT-8, an O-methylated isoflavone, called pratensein, and UM-9, a tri-O-methylated isoflavan, also called duartin, are isolated from Dalbergia parviflora, a plant native to tropical countries like Myanmar, Thailand, Malaysia, and Indonesia. Using HEK293 cells overexpressed TPC2 or TRPML1, Netcharoensirisuk et al.135 revealed the specific inhibitory effects of both compounds on TPC2 but not TRPML1. Both compounds were also shown to be more potent than naringenin (IC50 (naringenin) = 74 ± 9 µM; IC50 (MT-8) = 2.6 ± 0.3 µM; IC50 (UM-9) = 9.5 ± 2.8). MT-8 and UM-9 significantly attenuated the proliferation, invasion, and migration of wt MNT-1 human melanoma cells. In contrast, no effect was observed on the proliferation, invasion, and migration of TPC2 KO MNT-1 cells. Collectively, these data confirmed the specificity of both compounds on TPC2.
TPCs antagonists and chemoresistance
Chemotherapy and targeted therapy are standard methods for tumor management. However, resistance development of malignant cells against the therapeutic agents consequently leads to the failure of the treatment. Mechanically, general mechanisms and drug-specific are associated with the development of tumor drug resistance136. Hence, chemoresistance is a critical factor that drives tumor relapse and cancer-related mortalities137. Chemoresistance enables cancer cells to survive in the presence of therapeutics. It is a significant challenge that oncology investigation looks for to understand and overcome. Multiple molecular mechanisms regarding the promotion of cancer cells survival and avoidance of apoptosis in response to commonly used chemotherapeutics have been recognized, including various sets of signaling pathways for promoting chemoresistance138. Ca2+signaling emerges to be a key contributor to the cytotoxic effects of chemotherapy. A large number of chemotherapeutic agents provoke rapid onset of cytosolic Ca2+ rise. Diverse chemotherapeutic agents depend upon a Ca2+signaling component for inducing neoplastic cell death. Thus, modulation of Ca2+signaling can (re)sensitize or increase the responsiveness of cancer cells to chemotherapeutics 139.The correlation of chemoresistance with Ca2+ channel activity has been recognized since the 80s.Several Ca2+ channels have been associated with cancer cell resistance, and the blockade of Ca2+ channels was correlated with the improvement of anticancer drug cytotoxicity139. More recent findings declared the implication of TPCs/Ca2+ machinery in chemoresistance. Novel inhibitors of TPCs, SG-094 and SG-005overcome cancer-chemoresistance by inhibiting the efflux transporter p-glycoproteins. P-glycoproteins are the main source of resistance to standard cancer therapeutics and treatment failure due to the enhanced efflux of cytotoxic drugs 140. Müller et al.56 demonstrated that inhibition of TPCs by a genetic-KO system or using pharmacological antagonists had boosted cancer cells’ sensitivity to chemotherapy. KO of TPC2 in VCR-R CEM cells significantly increases the sensitivity of the cells to vincristine compared to the wt. The proliferation of TPC2 KO VCR-R CEM cells is greatly reduced in the presence of a lower concentration of vincristine (IC50wt: 3.3 µM, IC50 KO: 1.6 µM, 72 h). Similarly, a lower concentration of vincristine is required to induce apoptosis in TPC2 KO VCR-R CEM cells (EC50wt: 3.0 µM, EC50 KO: 1.7 µM, 48 h). Moreover, combination treatment of vincristine (0.01 and 0.1 µM) with tetrandrine, SG-94 or SG-005 (1 and 5 µM) synergistically enhanced treatment response of the wt VCR-R CEM cells and B-cell acute lymphoblastic leukemia (B-ALL) patient-derived xenograft (PDX) cells from a relapse patient. Hence, VCR-R CEM cells’ chemoresistance toward vincristine is effectively reversed by using vincristine and TPCs inhibitors as a combination therapy. Furthermore, TPC2 inhibitors including, well known antagonist tetrandrine, and novel agents SG-094 and SG-005 markedly sensitized sorafenib-resistant liver tumor cells and elevated the sensitivity of sorafenib-resistant liver tumor cells to sorafenib. Seco-analogs SG-094, and SG-005 compared to tetrandrine possessed lower toxicity to PBMCs and non-malignant hepatocytes. Hence, these seco-analogs potentially form novel and safealternatives and could be an effective strategy to reverse multidrug resistance in cancer 8.
Conclusion
This review summarizes the leading roles of TPCs/Ca2+signaling machinery in multiple cancer hallmarks, namely, cell proliferation, angiogenesis, migration, invasion, metastasis, and metabolic reprogramming. Furthermore, it demonstrates TPCs connection with chemoresistance and autophagy in cancer. It shows the intersections between TPCs and cancer patients’ survival which identifies TPCs as cancer biomarkers and cancer drivers. Additionally, pharmacological inhibitors of TPCs were also discussed, suggesting the potential of TPCs as a pharmacological target for various diseases, including cancer. Further research in this field is required to understand the exact mechanisms of the role of TPCs in cancer. Thorough mechanism-based pharmacology, metabolism, and pharmacokinetic evaluation on pharmacological inhibitors of TPCs are also warranted. Additionally, comprehensive toxicity assessments, especially long-term toxicity; in in vivo studies need to be performed. This includes the safety and toxicity evaluation of these agents for use in humans.
Acknowledgement
The authors acknowledge the Ministry of Higher Education Malaysia (MoHE) and UniversitiTeknologi MARA (UiTM) for funding this research through the Fundamental Research Grant Scheme (FRGS) (600-IRMI/FRGS 5/3 (315/2019)) and Lestari (600-IRMI/FRGS 5/3/LESTARI (014/2019)). The authors also acknowledge the Higher Education Development Program (HEDP) of the Ministry of Higher Education of Afghanistan and the Faculty of Pharmacy, Kabul University for supporting this study.
Funding Source
This work was funded by MoHE (600-IRMI/FRGS 5/3 (315/2019)) and UiTM (600-IRMI/FRGS 5/3/LESTARI (014/2019)).
Conflict of Interest
Declared none
References
- Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians. 2021;71(3):209-249. doi:10.3322/caac.21660
CrossRef - Luan F, He X, Zeng N. Tetrandrine: a review of its anticancer potentials, clinical settings, pharmacokinetics and drug delivery systems. Journal of Pharmacy and Pharmacology. 2020;72(11):1491-1512. doi:10.1111/jphp.13339
CrossRef - Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
CrossRef - Dowd J, Hendin J, Fukushiro-Lopes DF, Laczynski D, Gentile S. Ion Channels in Breast Cancer: From Signaling to Therapy. Breast Cancer – From Biology to Medicine. 2017;(April). doi:10.5772/66172
CrossRef - Kondratskyi A, Yassine M, Kondratska K, Skryma R, Slomianny C. Calcium-permeable ion channels in control of autophagy and cancer.Frontiers in Physiology. 2013;4(272):1-12. doi:10.3389/fphys.2013.00272
CrossRef - Faris P, Pellavio G, Ferulli F, et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca2+Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers (Basel). 2019;11(4):542.doi:10.3390/cancers11040542
CrossRef - Mathew M, Mateti UV, Saj N, Philip ML, Shetty V. Drug utilization evaluation of anticancer drugs in a charitable hospital. Indian Journal of Medical and Paediatric Oncology. 2019;40(1):105-110. doi:10.4103/ijmpo.ijmpo_156_18
CrossRef - Müller M. Novel Chemical Tools to Target Two-Pore Channel 2 , P-Glycoprotein and Histone Deacetylase 6 in Cancer. 2020.
- Prevarskaya N, Skryma R, Shuba Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiological Reviews. 2018;98(2):559-621. doi:10.1152/physrev.00044.2016
CrossRef - Capiod T, Shuba Y, Skryma R, Prevarskaya N. Calcium signalling and cancer cell growth. Subcell Biochem. 2007;45:405-427. doi:10.1007/978-1-4020-6191-2_15
CrossRef - Tedeschi V, Petrozziello T, Secondo A. Calcium Dyshomeostasis and Lysosomal Ca2+Dysfunction in Amyotrophic Lateral Sclerosis. Cells. 2019;8(10):1216.doi:10.3390/cells8101216
CrossRef - Clapham DE. Calcium Signaling.Cell. 2007;131(6):1047-1058.doi:10.1016/j.cell.2007.11.028
CrossRef - Tsai FC, Kuo GH, Chang SW, Tsai PJ. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. BioMed Research International. 2015. doi:10.1155/2015/409245
CrossRef - Monteith GR, Davis FM, Roberts-thomson SJ. Calcium Channels and Pumps in Cancer : Changes and Consequences. J Biol Chem. 2012;287(38):31666-31673.doi:10.1074/jbc.R112.343061
CrossRef - Parkash J, Asotra K. Calcium wave signaling in cancer cells. Life Sciences. 2010;87(19-22):587-595. doi:10.1016/j.lfs.2010.09.013
CrossRef - Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharmaceutica Sinica B. 2017;7(1):3-17. doi:10.1016/j.apsb.2016.11.001
CrossRef - Sharma V, Rana R, Baksi R, Borse SP, Nivsarkar M. Light-controlled calcium signalling in prostate cancer and benign prostatic hyperplasia. Future Journal of Pharmaceutical Sciences. 2020;6(1). doi:10.1186/s43094-020-00046-w
CrossRef - Bong AHL, Monteith GR. Calcium signaling and the therapeutic targeting of cancer cells. Biochimica et Biophysica Acta – Molecular Cell Research. 2018;1865(11):1786-1794. doi:10.1016/j.bbamcr.2018.05.015
CrossRef - Alharbi AF, Parrington J. Endolysosomal Ca2+ Signaling in Cancer: The Role of TPC2, From Tumorigenesis to Metastasis. Frontiers in Cell and Developmental Biology. 2019;7(302):1-7. doi:10.3389/fcell.2019.00302
CrossRef - Lloyd-evans E, Waller-evans H. Lysosomal Ca 2+ Homeostasis and Signaling in Health and Disease.Cold Spring Harb Perspect Biol.2020;12(6):a035311. doi:10.1101/cshperspect.a035311
CrossRef - Hu YB, Dammer EB, Ren RJ, Wang G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Translational Neurodegeneration. 2015;4(1):1-10. doi:10.1186/s40035-015-0041-1
CrossRef - Albrecht T, Zhao Y, Nguyen TH, Campbell RE, Johnson JD. Fluorescent biosensors illuminate calcium levels within defined beta-cell endosome subpopulations. Cell Calcium. 2015;57(4):263-274. doi:10.1016/j.ceca.2015.01.008
CrossRef - Galione A, Morgan AJ, Arredouani A, et al. NAADP as an intracellular messenger regulating lysosomal calcium-release channels.Biochem Soc Trans. 2010;38(6):1424-1431. doi:10.1042/BST0381424
CrossRef - Kintzer AF, Stroud RM. On the structure and mechanism of two-pore channels. FEBS Journal. 2018;285(2):233-243. doi:10.1111/febs.14154
CrossRef - Grimm C, Chen CC, Wahl-Schott C, Biel M. Two-pore channels: Catalyzers of endolysosomal transport and function. Frontiers in Pharmacology. 2017;8(45):6-11. doi:10.3389/fphar.2017.00045
CrossRef - Dickinson MS, Myasnikov A, Eriksen J, Poweleit N, Stroud RM. Resting state structure of the hyperdepolarization activated two-pore channel 3. Proceedings of the National Academy of Sciences of the United States of America. 2020;117(4):1988-1993. doi:10.1073/pnas.1915144117
CrossRef - Lagostena L, Festa M, Pusch M, Carpaneto A. The human two-pore channel 1 is modulated by cytosolic and luminal calcium. Scientific Reports. 2017;7:1-11. doi:10.1038/srep43900
CrossRef - Sakurai Y, Kolokoltsov AA, Chen C-c, et al. Targets for Disease Treatment. Science. 2015;347(6225):995-998.
CrossRef - Pitt SJ, Reilly-O’Donnell B, Sitsapesan R. Exploring the biophysical evidence that mammalian two-pore channels are NAADP-activated calcium-permeable channels. Journal of Physiology. 2016;594(15):4171-4179. doi:10.1113/JP270936
CrossRef - Cang C, Zhou Y, Navarro B, et al. MTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell. 2013;152(4):778-790. doi:10.1016/j.cell.2013.01.023
CrossRef - Wang X, Zhang X, Dong XP, et al. TPC proteins are phosphoinositide- Activated sodium-selective ion channels in endosomes and lysosomes. Cell. 2012;151(2):372-383. doi:10.1016/j.cell.2012.08.036
CrossRef - Pitt SJ, Funnell TM, Sitsapesan M, et al. TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+. Journal of Biological Chemistry. 2010;285(45):35039-35046. doi:10.1074/jbc.M110.156927
CrossRef - Pitt SJ, Lam AKM, Rietdorf K, Galione A, Sitsapesan R. Reconstituted Human TPC1 is a proton-permeable ion channel and is activated by NAADP or Ca2+. Science Signaling. 2014;7(326):1-12. doi:10.1126/scisignal.2004854
CrossRef - Rybalchenko V, Ahuja M, Coblentz J, et al. Membrane potential regulates Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) dependence of the pH- and Ca2+-sensitive organellar two-pore channel TPC1. Journal of Biological Chemistry. 2012;287(24):20407-20416. doi:10.1074/jbc.M112.359612
CrossRef - Gómez-Suaga P, Luzón-Toro B, Churamani D, et al. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Human Molecular Genetics. 2012;21(3):511-525. doi:10.1093/hmg/ddr481
CrossRef - Feijóo-Bandín S, García-Vence M, García-Rúa V, et al. Two-pore channels (TPCs): Novel voltage-gated ion channels with pleiotropic functions. Channels. 2017;11(1):20-33. doi:10.1080/19336950.2016.1213929
CrossRef - Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S. Convergent regulation of the lysosomal two-pore channel-2 by Mg 2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO Journal. 2014;33(5):501-511. doi:10.1002/embj.201387035
CrossRef - Zhang X, Chen W, Li P, et al. Agonist-specific voltage-dependent gating of lysosomal two-pore na+ channels. eLife. 2019;8:1-18. doi:10.7554/eLife.51423
CrossRef - D’amore A, Hanbashi AA, Di Agostino S, et al. Loss of two-pore channel 2 (TPC2) expression increases the metastatic traits of melanoma cells by a mechanism involving the hippo signalling pathway and store-operated calcium entry. Cancers. 2020;12(9):1-18. doi:10.3390/cancers12092391
CrossRef - Favia A, Pafumi I, Desideri M, et al. NAADP-Dependent Ca2+ Signaling Controls Melanoma Progression, Metastatic Dissemination and Neoangiogenesis. Scientific Reports. 2016;6(18925):1-12. doi:10.1038/srep18925
CrossRef - Nam O, Nguyen P, Grimm C, et al. Two-Pore Channel Function Is Crucial for the Migration of Invasive Cancer Cells. Cancer Res.2017;77(6):1427-1439. doi:10.1158/0008-5472.CAN-16-0852
CrossRef - Brailoiu E, Churamani D, Cai X, et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. Journal of Cell Biology. 2009;186(2):201-209. doi:10.1083/jcb.200904073
CrossRef - Davis LC, Morgan AJ, Chen J-l, et al. Report NAADP Activates Two-Pore Channels on T Cell Cytolytic Granules to Stimulate Exocytosis and Killing. Current Biology. 2012;22(24):2331-2337. doi:10.1016/j.cub.2012.10.035
CrossRef - Grimm C, Holdt LM, Chen CC, et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nature Communications. 2014;5(4699). doi:10.1038/ncomms5699
CrossRef - Ruas M, Rietdorf K, Arredouani A, et al. Purified TPC Isoforms Form NAADP Receptors with Distinct Roles for Ca2+ Signaling and Endolysosomal Trafficking. Current Biology. 2010;20(8):703-709. doi:10.1016/j.cub.2010.02.049
CrossRef - Ruas M, Chuang KT, Davis LC, et al. TPC1 Has Two Variant Isoforms, and Their Removal Has Different Effects on Endo-Lysosomal Functions Compared to Loss of TPC2. Molecular and Cellular Biology. 2014;34(21):3981-3992. doi:10.1128/mcb.00113-14
CrossRef - Ambrosio AL, Boyle JA, Aradi AE, Christian KA, Di Pietro SM. TPC2 controls pigmentation by regulating melanosome pH and size. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(20):5622-5627. doi:10.1073/pnas.1600108113
CrossRef - Jin X, Zhang Y, Alharbi A, Hanbashi A, Alhoshani A, Parrington J. Targeting Two-Pore Channels: Current Progress and Future Challenges. Trends in Pharmacological Sciences. 2020;41(8):582-594. doi:10.1016/j.tips.2020.06.002
CrossRef - Patel S, Kilpatrick BS. Two-pore channels and disease. Biochimica et Biophysica Acta – Molecular Cell Research. 2018;1865(11):1678-1686. doi:10.1016/j.bbamcr.2018.05.004
CrossRef - Parrington J, Lear P, Hachem A. Calcium signals regulated by NAADP and two-pore channels -their role in development, differentiation and cancer. International Journal of Developmental Biology. 2015;59(7-9):327-340. doi:10.1387/ijdb.150211jp
CrossRef - Gunaratne GS, Yang Y, Li F, Walseth TF, Marchant JS. NAADP-dependent Ca2+ signaling regulates Middle East respiratory syndrome-coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium. 2018;75:30-41. doi:10.1016/j.ceca.2018.08.003
CrossRef - Dobson SJ, Mankouri J, Whitehouse A, A requirement for Potassium and Calcium Channels during the Endosomal Trafficking of Polyomavirus Virions. bioRxiv. 2019;814681
CrossRef - Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nature Communications. 2020;11(1620). doi:10.1038/s41467-020-15562-9
CrossRef - Heister PM, Poston RN. Pharmacological hypothesis: TPC2 antagonist tetrandrine as a potential therapeutic agent for COVID-19. Pharmacology Research and Perspectives. 2020;8(5):1-8. doi:10.1002/prp2.653
CrossRef - Bahari NN, Jamaludin SYN, Jahidin AH. Assessment of TRPV4 Channel and Its Role in Colorectal Cancer Cells. Biomed Pharmacol J. 2019;12(2):629-638.
CrossRef - Müller M, Geisslinger F, Gerndt S, et al. Blocking Lysosomal Two-Pore Channel 2 Function Inhibits Proliferation of Multidrug Resistant Leukemia Cells and Sensitizes Them to Vincristine Treatment. Blood. 2019;134(Supplement_1):2081-2081. doi:10.1182/blood-2019-128195
CrossRef - Huang X, Godfrey TE, Gooding WE, McCarty KS Jr, Gollin SM. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer. 2006;45(11):1058-1069. doi:10.1002/gcc.20371
CrossRef - Faris P, Shekha M, Montagna D, Guerra G, Moccia F. Endolysosomal Ca2+Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind!. Cancers (Basel). 2018;11(1):27. doi:10.3390/cancers11010027
CrossRef - Grimm C, Bartel K, Vollmar AM, Biel M. Endolysosomal cation channels and cancer—a link with great potential. Pharmaceuticals. 2018;11(1):1-8. doi:10.3390/ph11010004
CrossRef - Sterea AM, Almasi S, El Hiani Y. The hidden potential of lysosomal ion channels: A new era of oncogenes. Cell Calcium. 2018;72:91-103. doi:10.1016/j.ceca.2018.02.006
CrossRef - Moccia F, Negri S, Shekha M, Faris P, Guerra G. Endothelial Ca2+ signaling, angiogenesis and vasculogenesis: Just what it takes to make a blood vessel. International Journal of Molecular Sciences. 2019;20(16):1-39. doi:10.3390/ijms20163962
CrossRef - Moccia F. Endothelial Ca2+ signaling and the resistance to anticancer treatments: Partners in crime. International Journal of Molecular Sciences. 2018;19(1). doi:10.3390/ijms19010217
CrossRef - Pafumi I, Festa M, Papacci F, et al. Naringenin Impairs Two-Pore Channel 2 Activity And Inhibits VEGF-Induced Angiogenesis.Sci Rep. 2017;7(1):1-11. doi:10.1038/s41598-017-04974-1
CrossRef - Favia A, Desideri M, Gambara G, Alessio AD, Ruas M, Esposito B. VEGF-induced neoangiogenesis is mediated by NAADPand two-pore channel-2-dependent Ca2+ signaling.Proc Natl Acad Sci U S A. 2014;111(44):E4706-15. doi:10.1073/pnas.1406029111
CrossRef - Varghese E, Samuel SM, Sadiq Z, et al. Anti-cancer agents in proliferation and cell death: The calcium connection. International Journal of Molecular Sciences. 2019;20(12). doi:10.3390/ijms20123017
CrossRef - Yu W, Yang L, Li T, Zhang Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Frontiers in Oncology. 2019;9. doi:10.3389/fonc.2019.00989
CrossRef - Farrag M, Anter A, Farrag N, Ibrahiem A. ‘Switch of E-Cadherin to N-Cadherin expression in different molecular subtypes of breast invasive duct carcinomas and its correlation with clinicopathological features’. Indian Journal of Pathology and Microbiology. 2021;64(1):38-46. doi:10.4103/IJPM.IJPM_924_19
- Horton JS, Wakano CT, Speck M, Stokes AJ. Two-pore channel 1 interacts with citron kinase, regulating completion of cytokinesis. Channels. 2015;9(1):21-29. doi:10.4161/19336950.2014.978676
CrossRef - Lin-Moshier Y, Keebler MV, Hooper R, et al. The Two-pore channel (TPC) interactome unmasks isoform-specific roles for TPCs in endolysosomal morphology and cell pigmentation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(36):13087-13092. doi:10.1073/pnas.1407004111
CrossRef - Bellono NW, Escobar IE, Oancea E. A melanosomal two-pore sodium channel regulates pigmentation. Scientific Reports. 2016;6:1-11. doi:10.1038/srep26570
CrossRef - Sun W, Yue J. TPC2 mediates autophagy progression and extracellular vesicle secretion in cancer cells. Experimental Cell Research. 2018;370(2):478-489. doi:10.1016/j.yexcr.2018.07.013
CrossRef - Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559-1564. doi:10.1126/science.1203543
CrossRef - Wang R-A, Lu Y-Y, Fan D-M. Reasons for cancer metastasis: A holistic perspective. Molecular and Clinical Oncology. 2015;3(6):1199-1202. doi:10.3892/mco.2015.623
CrossRef - Tajbakhsh A, Pasdar A, Rezaee M, et al. The current status and perspectives regarding the clinical implication of intracellular calcium in breast cancer. vol 233. 2018:5623-5641.
CrossRef - Boopathy GTK, Hong W. Role of Hippo Pathway-YAP/TAZ signaling in angiogenesis. Frontiers in Cell and Developmental Biology. 2019;7:1-12. doi:10.3389/fcell.2019.00049
CrossRef - Warren JSA, Xiao Y, Lamar JM. YAP/TAZ activation as a target for treating metastatic cancer. Cancers. 2018;10(4). doi:10.3390/cancers10040115
CrossRef - Ohshima K, Morii E. Metabolic reprogramming of cancer cells during tumor progression and metastasis. Metabolites. 2021;11(1):1-23. doi:10.3390/metabo11010028
CrossRef - Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368(6487). doi:10.1126/science.aaw5473
CrossRef - Sun L, Suo C, Li St, Zhang H, Gao P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochimica et Biophysica Acta – Reviews on Cancer. 2018;1870(1):51-66. doi:10.1016/j.bbcan.2018.06.005
CrossRef - Bruce JIE, James AD. Targeting the calcium signalling machinery in cancer. Cancers. 2020;12(9):1-34. doi:10.3390/cancers12092351
CrossRef - Müller M, Gerndt S, Chao Y-K, et al. Gene editing and synthetically accessible inhibitors reveal role for TPC2 in HCC cell proliferation and tumor growth. Cell Chemical Biology. 2021:3-3. doi:10.1016/j.chembiol.2021.01.023
CrossRef - Huang T, Song X, Yang Y, et al. Autophagy and hallmarks of cancer. Critical Reviews in Oncogenesis. 2018;23(5-6):247-267. doi:10.1615/CritRevOncog.2018027913
CrossRef - López-Pérez Ó, Badiola JJ, Bolea R, Ferrer I, Llorens F, Martín-Burriel I. An Update on Autophagy in Prion Diseases. Frontiers in Bioengineering and Biotechnology. 2020;8:1-15. doi:10.3389/fbioe.2020.00975
CrossRef - Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes and Development. 2011;25(5):460-470. doi:10.1101/gad.2016311
CrossRef - Strohecker AM, White E. Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy. 2014;10(2):384-385. doi:10.4161/auto.27320
CrossRef - Su Z, Li G, Liu C, et al. Autophagy inhibition impairs the epithelial-mesenchymal transition and enhances cisplatin sensitivity in nasopharyngeal carcinoma. Oncology Letters. 2017;13(6):4147-4154. doi:10.3892/ol.2017.5963
CrossRef - Kim YH, Baek SH, Kim EK, et al. Uncoordinated 51-like kinase 2 signaling pathway regulates epithelial-mesenchymal transition in A549 lung cancer cells. FEBS Letters. 2016;590(9):1365-1374. doi:10.1002/1873-3468.12172
CrossRef - Shen H, Yin L, Deng G, et al. Knockdown of Beclin-1 impairs epithelial-mesenchymal transition of colon cancer cells. Journal of Cellular Biochemistry. 2018;119(8):7022-7031. doi:10.1002/jcb.26912
CrossRef - Li J, Yang B, Zhou Q, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis. 2013;34(6):1343-1351. doi:10.1093/carcin/bgt063
CrossRef - Han C, Sun B, Wang W, et al. Overexpression of microtubule-associated protein-1 light chain 3 is associated with melanoma metastasis and vasculogenic mimicry. Tohoku Journal of Experimental Medicine. 2011;223(4):243-251. doi:10.1620/tjem.223.243
CrossRef - Zhao H, Yang M, Zhao J, Wang J, Zhang Y, Zhang Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Medical Oncology. 2013;30(1). doi:10.1007/s12032-013-0475-1
CrossRef - Sweatt SKGBACAYLYLL. 乳鼠心肌提取 HHS Public Access. Physiology & behavior. 2016;176(1):139-148. doi:10.1158/1078-0432.CCR-11-1282.Punctat
- Kroemer G, Mariño G, Levine B. Autophagy and the Integrated Stress Response. Molecular Cell. 2010;40(2):280-293. doi:10.1016/j.molcel.2010.09.023
CrossRef - Singh SS, Vats S, Chia AYQ, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37(9):1142-1158. doi:10.1038/s41388-017-0046-6
CrossRef - Kimmelman AC, White E. Autophagy and Tumor Metabolism. Cell Metabolism. 2017;25(5):1037-1043. doi:10.1016/j.cmet.2017.04.004
CrossRef - Dragowska WH, Weppler SA, Wang JC, et al. Induction of Autophagy Is an Early Response to Gefitinib and a Potential Therapeutic Target in Breast Cancer. PLoS ONE. 2013;8(10):1-20. doi:10.1371/journal.pone.0076503
CrossRef - Chen Y, Li X, Guo L, et al. Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Molecular medicine reports. 2015;12(2):1645-1652. doi:10.3892/mmr.2015.3623
CrossRef - Tseng HC, Liu WS, Tyan YS, Chiang HC, Kuo WH, Chou FP. Sensitizing effect of 3-methyladenine on radiation-induced cytotoxicity in radio-resistant HepG2 cells in vitro and in tumor xenografts. Chemico-Biological Interactions. 2011;192(3):201-208. doi:10.1016/j.cbi.2011.03.011
CrossRef - Chittaranjan S, Bortnik S, Dragowska WH, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clinical Cancer Research. 2014;20(12):3159-3173. doi:10.1158/1078-0432.CCR-13-2060
CrossRef - Golden EB, Cho HY, Jahanian A, et al. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurgical Focus. 2014;37(6):1-11. doi:10.3171/2014.9.FOCUS14504
CrossRef - Li J, Hou N, Faried A, Tsutsumi S, Kuwano H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. European Journal of Cancer. 2010;46(10):1900-1909. doi:10.1016/j.ejca.2010.02.021
CrossRef - Zhao XG, Sun RJ, Yang XY, et al. Chloroquine-enhanced efficacy of cisplatin in the treatment of hypopharyngeal carcinoma in xenograft mice. PLoS ONE. 2015;10(4):1-12. doi:10.1371/journal.pone.0126147
CrossRef - Das CK, Mandal M, Kögel D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer and Metastasis Reviews. 2018;37(4):749-766. doi:10.1007/s10555-018-9727-z
CrossRef - Janji B, Berchem G, Chouaib S. Targeting autophagy in the tumor microenvironment: New challenges and opportunities for regulating tumor immunity. Frontiers in Immunology. 2018;9:1-9. doi:10.3389/fimmu.2018.00887
CrossRef - Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479-483. doi:10.1038/nature19084
CrossRef - Sun WL, Chen J, Wang YP, Zheng H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy. 2011;7(9):1035-1044. doi:10.4161/auto.7.9.16521
CrossRef - Chen X, Wang P, Guo F, et al. Autophagy enhanced the radioresistance of non-small cell lung cancer by regulating ROS level under hypoxia condition. International Journal of Radiation Biology. 2017;93(8):764-770. doi:10.1080/09553002.2017.1325025
CrossRef - Ding ZB, Hui B, Shi YH, et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clinical Cancer Research. 2011;17(19):6229-6238. doi:10.1158/1078-0432.CCR-11-0816
CrossRef - Mohan N, Chakrabarti M, Banik NL, Ray SK. Combination of LC3 shRNA Plasmid Transfection and Genistein Treatment Inhibited Autophagy and Increased Apoptosis in Malignant Neuroblastoma in Cell Culture and Animal Models. PLoS ONE. 2013;8(10):1-16. doi:10.1371/journal.pone.0078958
CrossRef - Ko A, Kanehisa A, Martins I, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death and Differentiation. 2014;21(1):92-99. doi:10.1038/cdd.2013.124
CrossRef - Selvakumaran M, Amaravadi RK, Vasilevskaya IA, O’Dwyer PJ. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clinical Cancer Research. 2013;19(11):2995-3007. doi:10.1158/1078-0432.CCR-12-1542
- Krogsaeter EK, Biel M, Wahl-Schott C, Grimm C. The protein interaction networks of mucolipins and two-pore channels. Biochimica et Biophysica Acta – Molecular Cell Research. 2019;1866(7):1111-1123. doi:10.1016/j.bbamcr.2018.10.020
- Lin PH, Duann P, Komazaki S, et al. Lysosomal two-pore channel subtype 2 (TPC2) regulates skeletal muscle autophagic signaling. Journal of Biological Chemistry. 2015;290(6):3377-3389. doi:10.1074/jbc.M114.608471
- Lu Y, Hao BX, Graeff R, Wong CWM, Wu WT, Yue J. Two pore channel 2 (TPC2) inhibits autophagosomal-lysosomal fusion by alkalinizing lysosomal pH. Journal of Biological Chemistry. 2013;288(33):24247-24263. doi:10.1074/jbc.M113.484253
- Pereira GJS, Antonioli M, Hirata H, et al. Glutamate induces autophagy via the two-pore channels in neural cells. 2017;8(8):12730-12740.
- Lee H, Kim JW, Kim DK, et al. Calcium channels as novel therapeutic targets for ovarian cancer stem cells. International Journal of Molecular Sciences. 2020;21(7). doi:10.3390/ijms21072327
- Thompson BJ. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. BioEssays. 2020;42(5):1-16. doi:10.1002/bies.201900162
- Shivakumar M, Lee Y, Bang L, Garg T, Sohn KA, Kim D. Identification of epigenetic interactions between miRNA and DNA methylation associated with gene expression as potential prognostic markers in bladder cancer. BMC Medical Genomics. 2017;10(Suppl 1)doi:10.1186/s12920-017-0269-y
- Li F, Ji JP, Xu Y, Liu RL. Identification a novel set of 6 differential expressed genes in prostate cancer that can potentially predict biochemical recurrence after curative surgery. Clinical and Translational Oncology. 2019;21(8):1067-1075. doi:10.1007/s12094-018-02029-z
- Penny CJ, Vassileva K, Jha A, et al. BBA – Molecular Cell Research Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers.Biochim Biophys Acta Mol Cell Res. 2019;1866(7):1151-1161. doi:10.1016/j.bbamcr.2018.10.022
- Naylor E, Arredouani A, Vasudevan SR, et al. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol.2009;5(4):220-226. doi:10.1038/nchembio.150
- Bellik Y, Boukraâ L, Alzahrani HA, et al. Molecular mechanism underlying anti-inflammatory and anti-Allergic activities of phytochemicals: An update. Molecules. 2013;18(1):322-353. doi:10.3390/molecules18010322
- Zhao H, Luo F, Li H, Zhang L, Yi Y, Wan J. Antinociceptive effect of tetrandrine on LPS-induced hyperalgesia via the inhibition of IKKβ phosphorylation and the COX-2/PGE2 pathway in mice. PLoS ONE. 2014;9(4):1-10. doi:10.1371/journal.pone.0094586
- Li X, Jin Q, Wu YL, et al. Tetrandrine regulates hepatic stellate cell activation via TAK1 and NF-κB signaling. International Immunopharmacology. 2016;36:263-270. doi:10.1016/j.intimp.2016.04.039
CrossRef - Gao S, Cui YL, Yu CQ, Wang QS, Zhang Y. Tetrandrine exerts antidepressant-like effects in animal models: Role of brain-derived neurotrophic factor. Behavioural Brain Research. 2013;238(1):79-85. doi:10.1016/j.bbr.2012.10.015
CrossRef - Jang BC. Tetrandrine has anti-adipogenic effect on 3T3-L1 preadipocytes through the reduced expression and/or phosphorylation levels of C/EBP-α, PPAR-γ, FAS, perilipin A, and STAT-3. Biochemical and Biophysical Research Communications. 2016;476(4):481-486. doi:10.1016/j.bbrc.2016.05.150
CrossRef - Mir IA, Tiku AB. Chemopreventive and therapeutic potential of “Naringenin,” a flavanone present in citrus fruits. Nutrition and Cancer. 2015;67(1):27-42. doi:10.1080/01635581.2015.976320
CrossRef - Li Q, Wang Y, Zhang L, et al. Naringenin exerts anti-angiogenic effects in human endothelial cells: Involvement of ERRα/VEGF/KDR signaling pathway. Fitoterapia. 2016;111:78-86. doi:10.1016/j.fitote.2016.04.015
CrossRef - Alam MA, Subhan N, Rahman MM, Uddin SJ, Reza HM, Sarker SD. Effect of citrus flavonoids, naringin and naringenin, on metabolic syndrome and their mechanisms of action. Advances in Nutrition. 2014;5(4):404-417. doi:10.3945/an.113.005603
CrossRef - Abaza MSI, Orabi KY, Al-Quattan E, Al-Attiyah RaJ. Growth inhibitory and chemo-sensitization effects of naringenin, a natural flavanone purified from Thymus vulgaris, on human breast and colorectal cancer. Cancer Cell International. 2015;15(1):1-19. doi:10.1186/s12935-015-0194-0
CrossRef - Sulfikkarali N, Krishnakumar N, Manoharan S, Nirmal RM. Chemopreventive efficacy of naringenin-loaded nanoparticles in 7,12-dimethylbenz(a)anthracene induced experimental oral carcinogenesis. Pathology and Oncology Research. 2013;19(2):287-296. doi:10.1007/s12253-012-9581-1
CrossRef - Leonardi T, Vanamala J, Taddeo SS, Davidson LA, Murphy ME, Patil BS, Wang N, Carroll RJ, Chapkin RS, Lupton JR, Turner ND. Apigenin and naringenin suppress colon carcinogenesis through the aberrant crypt stage in azoxymethane-treated rats. Exp Biol Med (Maywood). 2010;235(6):710-7. doi: 10.1258/ebm.2010.009359.
CrossRef - D’Amore A, Gradogna A, Palombi F, et al. The Discovery of Naringenin as Endolysosomal Two-Pore Channel Inhibitor and Its Emerging Role in SARS-CoV-2 Infection. Cells. 2021;10(5):1130-1130. doi:10.3390/cells10051130
CrossRef - Clementi N, Scagnolari C, D’Amore A, et al. Naringenin is a powerful inhibitor of SARS-CoV-2 infection in vitro. Pharmacol Res. 2021;163:105255. doi:10.1016/j.phrs.2020.105255
CrossRef - Netcharoensirisuk P, Abrahamian C, Tang R, et al. Flavonoids increase melanin production and reduce proliferation, migration and invasion of melanoma cells by blocking endolysosomal/melanosomal TPC2. Scientific Reports. 2021;11(1):1-14. doi:10.1038/s41598-021-88196-6
CrossRef - Chen X, Chen S, Yu D. Metabolic reprogramming of chemoresistant cancer cells and the potential significance of metabolic regulation in the reversal of cancer chemoresistance. Metabolites. 2020;10(7):1-15. doi:10.3390/metabo10070289
CrossRef - Madden EC, Gorman AM, Logue SE, Samali A. Tumour Cell Secretome in Chemoresistance and Tumour Recurrence. Trends in Cancer. 2020;6(6):489-505. doi:10.1016/j.trecan.2020.02.020
CrossRef - Yeldag G, Rice A, Hernández AdR. Chemoresistance and the self-maintaining tumor microenvironment. Cancers. 2018;10(12)doi:10.3390/cancers10120471
CrossRef - Kerkhofs M, Bittremieux M, Morciano G, et al. Emerging molecular mechanisms in chemotherapy: Ca2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death and Disease. 2018;9(3). doi:10.1038/s41419-017-0179-0
CrossRef - Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Scientific Reports. 2018;8(1):1-18. doi:10.1038/s41598-018-19325-x
CrossRef

