
Manuscript accepted on :11-Mar-2021
Published online on: 26-03-2021
Plagiarism Check: Yes
Reviewed by: Dr. Tsvetelina Velikova
Second Review by: Dr. Ankur Singh Bist
Final Approval by: Dr. H Fai Poon
Semuel Sandy* and Irawaty Wike
Papua Health Research and Development Institute-Indonesia Ministry of Health, Indonesia.
Corresponding Author E-mail: mercury.sandy56@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/2144
Abstract
Purpose: In this review, the compound 6-prenylapigenin was identified as a potential wild type Plasmodium vivax dihydrofolate reductase (PDB ID: 2BL9) protein receptor inhibitor through a series of computer-assisted drug design processes, to highlight important interactions between ligand and 2BL9 receptor protein and determine drug properties. proposed as a 2BL9 inhibiting agent.
Methods: The in silico study used secondary data including Plasmodium vivax protein receptor (PDB ID: 2BL9), 6-Prenylapigenin compound (PubChem ID: 10382485), and native ligand Pyrimethamine (PubChem ID: 4993) as a comparison. In silico analysis using software, including AutoDock v 4.2.3, admetSAR v 2.0, Lipinski Role Of Five, PROCHECK SAVES v 6.0, LigPlus + v 2.2 and the Discovery Studio 2016.
Results: The study results showed that the free energy of the Gibbs bonding compound 6-Prenylapigenin is -7.61 kcal/mol with an inhibition constant is 2.65 nM. Types of hydrogen bonding to the amino acid residues Asp53 (A) and Ile173 (A). Hydrophobic extraction of the amino acid residues were Tyr125 (A); Met54 (A); Leu128 (A); Phe57 (A); Ala15 (A); Cys14 (A); Leu39 (A); Leu45 (A); and Tyr179 (A). In silico studies, this compound also has good toxicity and bioavailability properties.
Conclusion: 6-Prenylapigenin compound has an inhibitor activity at the active site of the 2BL9 protein receptor by forming hydrogen bonding and hydrophobic interactions. This compound has good toxicity and bio availability so that it may be developed as a dihydrofolate reductase enzyme inhibitor compound.
Keywords
Antimalarial; Cannabis sativa L; DHFR; In-Silico; Plasmodium vivax
Download this article as:| Copy the following to cite this article: Sandy S, Wike I. In Silico Antimalarial 5,7-Dihydroxy-2- (4-Hydroxyphenyl) -6- (3-Methylbut-2-Enyl) Chromen-4-one (6-Prenylapigenin) Plant Cannabis Sativa L. (Cannabaceae) Enzyme Inhibitor of DHFR Plasmodium Vivax.Biomed Pharmacol J 2021;14(1). |
| Copy the following to cite this URL: Sandy S, Wike I. In Silico Antimalarial 5,7-Dihydroxy-2- (4-Hydroxyphenyl) -6- (3-Methylbut-2-Enyl) Chromen-4-one (6-Prenylapigenin) Plant Cannabis Sativa L. (Cannabaceae) Enzyme Inhibitor of DHFR Plasmodium Vivax.Biomed Pharmacol J 2021;14(1). Available from: https://bit.ly/3d9VIS9 |
Introduction
Malaria is caused by infection with the apicomplexan parasite of the genus Plasmodium. An estimated 219 million cases and 405,000 deaths from malaria occurred in 2018 alone, with children under 5 representing two-thirds of these incidents 1. Resistance to available antimalarials, particularly to WHO recommended artemisinin combination therapy (ACTs), has falciparum (Pf) malaria were reported in 2004 in Cambodia and raised concerns because no alternative first-line treatment was available. 34. Recurrence resistance, coupled with a slowly increasing incidence rate (2% since 2015) means that new anti-malarial designs need to be implemented efficiently and rationally 5.
Cannabis sativa L. (Cannabaceae) is a widespread species. This plant is found in various habitats, including coastal areas, temperate valley areas, and alpine mountains in the Himalayas. Marijuana has a long history of medicinal use in the Middle East and Asia dating back to the 6th century BC 6. This herb was introduced in Western Europe as a medicine in the early 19th century to treat epilepsy, tetanus, rheumatism, migraine, asthma, trigeminal neuralgia, fatigue, and insomnia. 7. This plant has the potential as an antiplasmodial because it contains 6- prenylapigenin at an inhibitory concentration of IC50 6.7 µM. 8,9.
This in silico study aims to study the molecular interaction of the 6-prenylapigenin compound as an antimalarial inhibitor of DHFR enzyme using Pyrimethamine as a comparison. This study used a crystal structure model of the Plasmodium vivax dihydrofolate reductase wild type protein (PDB ID: 2BL9). Dihydrofolate reductase enzymes have an important role in cell growth and proliferation by catalyzing the reduction of dihydrofolic acid to tetrahydrofolic acid. Tetrahydrofolic acid is a derivative of folic acid (folate) and is needed by Plasmodium spp for the synthesis of deoxythymidine monophosphate, a DNA monomer. 7-10.
Materials and Methods
The hardware used by the Azus 3FBS7F0J Laptop is an intel ® Celeron ® CPU N3350 @ 1.10 GHz, 4 GB RAM, 64 bit operating system, x64 based processor. The software used includes: admetSAR V.2.0 (http://lmmd.ecust.edu.cn/ admetsar2); Lipinski Role Of Five (http://www.scfbio-iitd.res.in/ software/ drugdesign/ lipinski.jsp#anchortag) Chimera v 1.11.2, PROCHECK SAVES v6.0 (https: // saves.mbi.ucla .edu / results? job = 600021 & p = procheck), AutoDock 4.2.3, AutoDock MGL Tools. 1.5.6; Discovery Studio 2016; Open Babel 2.4.1; LigPlus ++ V2.2
Preparation of Ligand
The test ligand is an antiplasmodial compound, a secondary metabolite compound from the Cannabis sativa L. (Cannabaceae) plant, namely 6-Prenylapigenin (5,7-dihydroxy-2- (4-hydroxyphenyl) -6- (3-methylbut-2-enyl) chromen-4 -one) downloaded on the site: PubChem (https://pubchem.ncbi.nlm.nih.gov/ compound/ 10382485# section= Names-and-Identifiers). The test ligands were prepared for structural optimization using Chimera v 1.11.2 software with the addition of hydrogen atoms, steepest descent steps 1000, steepest descent size 0.02 Å, conjugate gradient steps 1000, conjugate gradient size 0.02 Å with intervals of 10, AMBERff14SB force fields with the charge structure of the Gasteiger method 11. The test ligands were analyzed using Lipinski Role Of Five (http://www.scfbio-iitd.res.in/ software/drugdesign/lipinski.jsp#anchortag) and analysis of absorption, distribution, metabolism, excretion and toxicity characteristics (ADMET) using admetSAR V.2.0 (http://lmmd.ecust.edu.cn/admetsar2)12.
Preparation of Protein
The protein receptor used as a target macromolecule is the crystal structure of the protein X-ray crystal structure of Plasmodium vivax dihydrofolate reductase wild type (PDB ID: 2BL9) with the parameters of the X-Ray Diffraction experimental method, Resolution: 1.90 ÅR-Value Free: 0.260 R-Value Work: 0.208 R-Value Observed: 0.208 (downloaded at https://www.rcsb.org/structure/2BL9). The receptor protein 2BL9 consists of chain A, CP6 ligands, and NDP. The protein optimization process uses Chimera v1.11.2 software. The objective of optimizing the protein model (energy minimization) is to optimize the conformation of the protein so that the optimal conformation is obtained 13. Analysis of protein structure stability using the Ramachandran Plot method using online software SAVES v6.0 (https://saves.mbi.ucla.edu/results?job=600021&p=procheck)14.
Docking validation
The structure of the Plasmodium vivax dihydrofolate reductase wild type (2BL9) crystal complex consisting of chain A, as well as atoms and other ligands. Proteins are prepared by removing other atoms, water, and unnecessary ligands are removed from the A-chain protein so as not to interfere with the binding process of the test ligand. The protein structure was optimized using Chimera 1.11.2 software with the addition of a hydrogen atom, the AMBERff14SB force fields parameter with the charge potential AM1 BCC 11,15. The optimized A-chain protein is stored in the data bank protein extension format (“*.pdb”). Native ligand derived from the protein A 2BL9 crystal complex is used for the binding validation process. Pyrimethamine (CP6) native ligand was separated using Chimere 1.11.2 software. The ligand obtained was structure optimized by adding parameters of hydrogen atoms, AMBERff14SB force fields, and charge potential of the Gastaiger method. The native ligand preparation results are stored in an extension format (“* .pdb”). The native ligand was redocking on protein A 2BL9 using AutoDock v.4.2.1 application. The grid box parameter used is the center ligand, with the grid box size x = 126; y = 126, and z = 126, spacing parameter = 0.100 Å, center grid box x = 89.467; y = 13,416; z = 33,996, while the docking parameters used by Lamarckian GA number of GA runs 50, population size 300, number of evals 2,500,000 (medium), maximum number of generation 27,00016. The results of the redocking protocol are valid if they have a Root Mean Square Deviation (RMSD) value ≤ 2 Å 11. Then these parameters are used as a protocol for docking using another test ligand.
Data analysis
Data analysis was performed based on the results of Gibbs binding energy (∆G) produced from ligands and proteins docking. The value of Gibbs free energy (∆G) indicates the strength of the bond between the test compound and the receptor. The lower the binding energy, the stronger the bond between the compound and the protein receptor 17.
Visualization of docking results
The interaction of the test ligand with the Protein A 2BL9 receptor was analyzed and interpreted using a two-dimensional visualization of the LigPlus + v 2.2 software and the Discovery Studio 2016 software. 16.
Results and Discussion
Analysis of the stability of the protein structure results from the optimization of the Plasmodium vivax dihydrofolate reductase wild type (PDB ID: 2BL9) protein using the Ramachandran Plot method can be seen in Figure 1. The Ramachandran Plot area is divided into four areas, namely the prohibited area (white color region), the allowed area (cream color region), the most favored area (red color region), the allowed region (yellow color region). Data of amino acid residues in the most favored regions were 89.2%, the additional allowed regions were 10.8%, the generously allowed regions were 0.0% and the disallowed regions were 0.0%. Disallowed regions were smaller than 15% and the smaller the percentage, the better the protein structure quality 18. The quality of the structure based on its geometry can be seen from the percentage of amino acid residues in the most favored and disallowed regions. A good structure is one that has the most favored regions percentage greater than 50%. The greater the percentage of amino acid residues that are in the most favored regions and the lower the percentage of residues in the disallowed region, the better the structural quality will be 18.
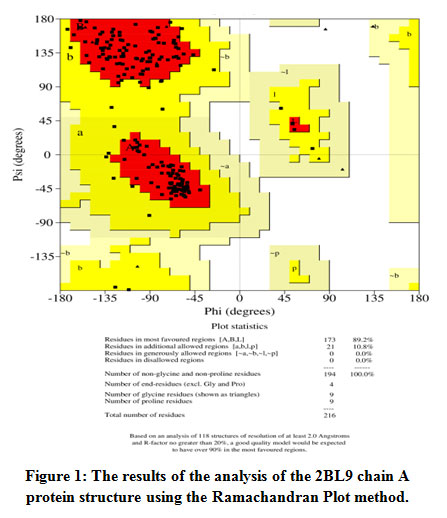 |
Figure 1: The results of the analysis of the 2BL9 chain A protein structure using the Ramachandran Plot method. |
The results of protein analysis showed that the structure of the 2BL9 protein chain A optimization results could be used as an in silico docking model as a target receptor. Before docking the ligand of the test compound, docking validation was carried out using the native ligand Pyrimethamine as the test ligand on 2BL9 chain A protein. Docking validation using the Root Mean Square Deviation (RMSD) parameter 19. The revoking results obtained an RMSD value of 0.96 Å (Figure 2). The validation protocol is successful if the RMSD value is obtained, namely the ratio of the atomic position between the experimental structure (reference ligand) and the structure that is blocked or predicted. The smaller the RMSD value, it shows that the predicted ligand pose is better because it approaches the native ligand conformation 20. The validation protocol used for docking is valid, so the docking validation protocol is used to test the test ligand for the 6-prenylapigenin compound.
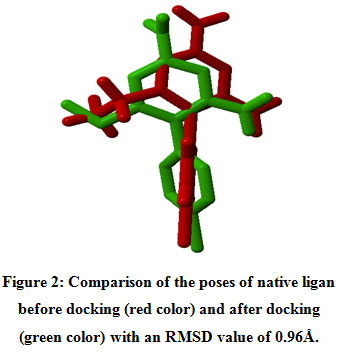 |
Figure 2: Comparison of the poses of native ligan before docking (red color) and after docking (green color) with an RMSD value of 0.96Å. |
The results of Pyrimethamine docking native ligand analysis obtained Gibbs binding energy (∆G) -7.26 kcal/mol and an inhibition constant (Ki) of 4.54 nM (Table 1). The free Gibbs binding energy (∆G) is a parameter of the stability of the conformation between the ligand and receptor. The ligand-receptor interactions will tend to be in the lowest energy state 15. This condition causes the molecule to be in a stable state so that the smaller the value of Gibbs binding energy (∆G), the interaction of the ligand with the receptor becomes more stable. 11. Hydrogen bonding formed from the interaction of the ligand native Pyrimethamine with the amino acid residue of 2BL9 chain A protein, namely Ile13 (A); Ile173 (A); Asp (53); Tyr179 (A). The same research by Choowongkomon. et.al 2010 mentioned the interaction of hydrogen pyrimethamine binding with the amino acid residues of the PvDHFR protein, including Ile13, Leu45, Asp53, Phe 57, Ile173, Thr194, Ser120. This difference is due to the docking parameters used and the two-dimensional visualization analysis method using the discovery studio application. The hydrogen bonding that is formed stabilizes the structure of the ligand-receptor interactions. Meanwhile, the hydrophobic interaction with the amino acid residues of Met54 (A); Cys14 (A); Ala15 (A); The Phe57 (A) formed helps to increase the stability of the hydrophobic bonds, minimizing the interaction of non-polar amino acid residues with water solvents (Figure 3) 17.
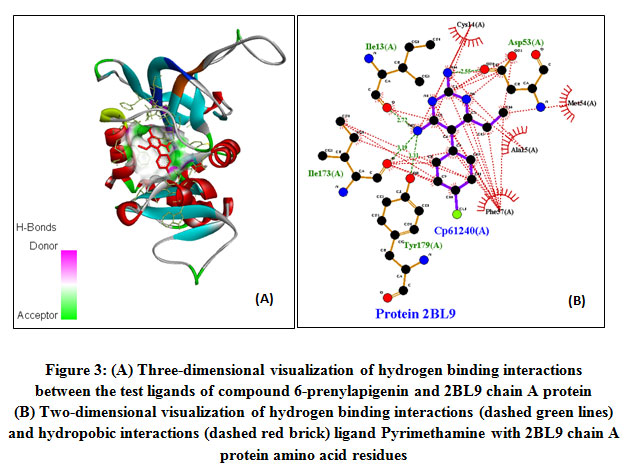 |
Figure 3: (A) Three-dimensional visualization of hydrogen binding interactions between the test ligands of compound 6-prenylapigenin and 2BL9 chain A protein. (B) Two-dimensional visualization of hydrogen binding interactions (dashed green lines) and hydropobic interactions (dashed red brick) ligand Pyrimethamine with 2BL9 chain A protein amino acid residues.Click here to view figure |
Tabel 1: The result of the addition of ligand native Pyrimethamine and ligand 6-prenylapigenin.
| Ligan | PubChem ID | Energy binding Gibbs (∆G)
(kcal/mol) |
KI
(nM) |
Hydrogen bindings | Hydrophobic interactions |
| 5-(4-Chloro-Phenyl)-6-Ethyl-Pyrimidine-2,4-Diamine (Pyrimethamine) | 4993 | -7.29 | 4.54 | Ile13(A); Ile173(A); Asp(53); Tyr179(A) | Met54(A); Cys14(A); Ala15(A); Phe57(A) |
| 5,7-dihydroxy-2-(4-hydroxyphenyl)-6-(3-methylbut-2-enyl)chromen-4-one (6-prenylapigenin) | 10382485 | -7.61 | 2.65 | Asp53(A); Ile173(A) | Tyr125(A); Met54(A); Leu128(A); Phe57(A); Ala15(A); Cys14(A); Leu39(A); Leu45(A); Tyr179(A) |
The result of ligand docking for the 6-prenylapigenin compound obtained the Gibbs bond energy of -7.61 kcal/mol with an inhibition constant in silico of 2.65 nM. When compared with native ligand Pyrimethamine, the free energy of Gibbs bonds (∆G) and the inhibition constant formed is lower, but the number of hydrogen bonds formed is less, among others, on the amino acid residue Asp53 (A) and Ile173 (A). the structural stability of the interaction of the 6-prenylapigenin ligand with the protein 2BL9 receptor was mostly due to the hydrophobic interaction of the amino acids residue Tyr125 (A); Met54 (A); Leu128 (A); Phe57 (A); Ala15 (A); Cys14 (A); Leu39 (A); Leu45 (A); and Tyr179 (A) (Figure 4). Hydrophobicity determines many biological processes, such as transport, distribution, metabolism, and molecular interactions of biological molecules. It was reported that binding affinity and drug efficacy could be optimized and increased by including hydrophobic groups 21,22
 |
Figure 4: (A) Three-dimensional visualization of hydrogen bonding interactions between the test ligands of compound 6-Prenylapigenin and 2BL9 chain A protein (B) Two-dimensional visualization of hydrogen bonding interactions (dashed green lines) and hydrophobic interactions (dashed red brick) ligands 6 -Prenylapigenin with 2BL9 chain A protein amino acid residues. |
Table 1 showed the results of the in silico analysis of the ligand 6-prenylapigenin test and the natural ligand Pyrimethamine fulfilling the Lipinski Role of Five criteria so that these compounds can be designed to be used orally. The results of in silico ADMET analysis showed that there was a small effect of AMES toxicity (mutagenic) and carcinogens. The absorption of these two compounds in the human intestine is very good with a probability of 99%. The Lipinski Rule of Five helps in differentiating between potentially drug-drug molecules that can be administered orally and those that cannot. This rule accurately predicts the likelihood of success or failure of a new oral drug candidate. Drug candidate compounds that do not meet any of these rules are not recommended for oral use designs due to poor absorption of the drug. The Lipinski Rule of Five is an exception for drugs that are given intravenously because they are not absorbed 23. Here’s the Lipinski Rule of Five:
Molecular weight ≤ 500 g / mol
Lipophilic (Log P <5)
Donor hydrogen atom (number of N and O atoms) <5
Hydrogen atomic acceptors (Number of OH and NH groups) <10
Molar reflection should be between 40-130
Additional criteria for determining the bioavailability of novel drug candidates for oral administration include:24
Total bond torque/rotation ≤ 10
Polar surface area of a compound (Polar surface area / TSA) ≤ 140Å2
Tabel 2: Results of Lipinski Role Five Analysis of Pyrimethamine and 6-Prenylapigenin.
| Ligands | PubChem ID | Molecular Weight (g/mol) | hydrogen bond donor | hydrogen bond acceptors | Log P | Molar Refractivity | Rotatable bonds | PSA (Å2) |
| 5-(4-Chloro-Phenyl)-6-Ethyl-Pyrimidine-2,4-Diamine (Pyrimethamine) | 4993 | 248.50 | 4 | 4 | 1.75 | 67.75 | 2 | 77.8 |
| 5,7-dihydroxy-2-(4-hydroxyphenyl)-6-(3-methylbut-2-enyl)chromen-4-one (6-prenylapigenin) | 10382485 | 338.00 | 3 | 5 | 3.92 | 93.94 | 3 | 87 |
Tabel 3 Showed that the results of in silico analysis of the 6-Prenylapigenin test ligand and the natural ligand Pyrimethamine meet the Lipinski Role of Five criteria so that these compounds can be designed to be used orally 22. The results of in silico ADMET analysis showed that there was a small effect of AMES toxicity (mutagenic) and carcinogens. The absorption of these two compounds in the human intestine is very good with a probability of 99%.
Tabel 3: Results of ADMET analysis of Pyrimethamine and 6-Prenylapigenin.
| Ligands | Pub Chem ID | AdmetSAR | ||||
| AMES toxicity (mutagenic) | Carcinogens | Blood-Brain Barrier | Human Intestinal Absorbtion | Toksisitas Akut (Rat)
LD50 (mol/Kg) |
||
| 5-(4-Chloro-Phenyl)-6-Ethyl-Pyrimidine-2,4-Diamine (Pyrimethamine) | 4993 | Non (0.9133) | Non (0.8016) | Yes (0.9383) | Yes (0.9973) | 2.7833 |
| 5,7-dihydroxy-2-(4-hydroxyphenyl)-6-(3-methylbut-2-enyl)chromen-4-one (6-prenylapigenin) |
10382485 |
Non (O.7708) | Non (0.9370) | Non(0.7023) | Yes (0.9959) |
2.9772 |
This study is still limited to in silicon testing using the docking method, laboratory experimental in vitro studies are needed to determine and require the interactions that occur in the 6-Prenylapigenin test compound with the amino acid residue of 2BL9 chain A protein.
Conclusion
The 6-Prenylapigenin compound test ligand has a lower bond-free energy and inhibition constant than the native ligand Pyrimethamine. So that the in silico prediction of these compounds has the same activity on the active site of the comparative ligand Pyrimethamine inhibits enzyme activity to synthesize folic acid in Plasmodium vivax.
Conflict of Interest
The authors declare that there are no conflicts of interest. Acknowledgment We are grateful to the “Association Marocaine des Chimistes Théoriciens” (AMCT) for its pertinent help concerning the programs.
References
- WHO. World Malaria Report 2019. WHO Global Malaria Programme and WHO Public Health Information and Geographic Systems; 2019.
- WHO. Global Technical Strategy for Malaria 2016-2030.; 2015.
- Dondorp AM, Yeung S, White L, et al. Artemisinin resistance: Current status and scenarios for containment. Nat Rev Microbiol. 2010;8(4):272-280. doi:10.1038/nrmicro2331
CrossRef - O’Brien C, Henrich PP, Passi N, Fidock DA. Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr Opin Infect Dis. 2011;24(6):570-577. doi:10.1097/QCO.0b013e32834cd3ed
CrossRef - Calic PPS, Mansouri M, Scammells PJ, McGowan S. Driving antimalarial design through understanding of target mechanism. Biochem Soc Trans. 2020;48(5):2067-2078. doi:10.1042/BST20200224
CrossRef - Andre CM, Hausman JF, Guerriero G. Cannabis sativa: The plant of the thousand and one molecules. Front Plant Sci. 2016;7(FEB2016):1-17. doi:10.3389/fpls.2016.00019
CrossRef - Chen MJ, Shimada T, Moulton AD, et al. The functional human dihydrofolate reductase gene. J Biol Chem. 1984;259(6):3933-3943. doi:10.1016/S0021-9258(17)43186-3
CrossRef - Soré H, Sanon S, Hilou A. Antiplasmodial properties of plants isolated flavonoids and their derivatives. Int J Herb Med. 2018;6(5):43-56.
- Radwan MM, Elsohly MA, Slade D, et al. Non-cannabinoid constituents from a high potency Cannabis sativa variety. Phytochemistry. 2016;69(14):2627-2633. doi:10.1016/j.phytochem.2008.07.010.
CrossRef - Bilsland E, Vliet L Van, Williams K, et al. Plasmodium dihydrofolate reductase is a second enzyme target for the antimalarial action of triclosan. 2018;:1-8. doi:10.1038/s41598-018-19549-x
CrossRef - Prieto-Martínez FD, Arciniega M, Medina-Franco JL. Acoplamiento Molecular: Avances Recientes y Retos. TIP Rev Espec en Ciencias Químico-Biológicas. 2018;21:65-87. doi:10.22201/fesz.23958723e.2018.0.143
CrossRef - Devvret, Pant K, Thapliyal A, Tufchi N. In Silico Docking analysis of Mycobacterium tuberculosis potential targets AftB and EmbA with selected phytochemicals. Int J Pharm Res Technol. 2019;7(2):15-22. doi:10.31838/ijprt/07.02.03
CrossRef - Lukitaningsih E, Mustikawaty AA, Sudarmanto A. Homology Modeling and Molecular Docking of Active Compounds from Bengkoang ( Pachyrrhizus erosus ) as Tyrosinase. J Ilmu Kefarmasian Indones. 2013;11(2):134-141.
- Suhadi A, Rizarullah R, Feriyani F. Simulasi Docking Senyawa Aktif Daun Binahong Sebagai Inhibitor Enzyme Aldose Reductase. Sel J Penelit Kesehat. 2019;6(2):55-65. doi:10.22435/sel.v6i2.1651
CrossRef - Pamudi BF, Azizahwati, Yanuar A. In-silico screening against antimalarial target plasmodium falciparum enoyl-acyl carrier protein reductase. Asian J Pharm Clin Res. 2017;10(Special Issue October):127-129. doi:10.22159/ajpcr.2017.v10s5.23114
CrossRef - Silva Andrade B, Ghosh P, Barh D, et al. Computational screening for potential drug candidates against the SARS-CoV-2 main protease. F1000Research. 2020;9:514. doi:10.12688/f1000research.23829.1
CrossRef - Ruslin, Syam Febriantara, Yamin. Studi In Silico Senyawa 2-amino-5-(3-(4-hydroxy-3,5-dimethoxy- benzoyl)guanidino)pentanoic acid dan Turunannya sebagai Inhibitor Phospodiesterase-5. Maj Farm Sains, dan Kesehat. 2014;2(1):22-26.
- Amelia F. Modeling Struktur Protein Vaksin H5N1 HA BTB Menggunakan I-Tasser. J Chem Inf Model. 2019;53(9):1689-1699.
- Jain AN, Nicholls A. Recommendations for evaluation of computational methods. J Comput Aided Mol Des. 2008;22(3-4):133-139. doi:10.1007/s10822-008-9196-5
CrossRef - Agista DD, Purnomo H, Tegar M, Nugroho AE. Interaction Between Active Compounds From Aegle marmelos Correa As Anti Inflammation Agent With Cox-1 And Cox-2 Receptor. Tradit Med J. 2015;18(2):80-87. doi:10.22146/tradmedj.7983
- Qian SB, Waldron L, Choudhary N, Klevit RE, Chazin WJ, Patterson C. Engineering a ubiquitin ligase reveals conformational flexibility required for ubiquitin transfer. J Biol Chem. 2009;284(39):26797-26802. doi:10.1074/jbc.M109.032334
CrossRef - Egieyeh SA, Syce J, Malan SF, Christoffels A. Prioritization of anti-malarial hits from nature: Chemo-informatic profiling of natural products with in vitro antiplasmodial activities and currently registered anti-malarial drugs. Malar J. 2016;15(1):1-23. doi:10.1186/s12936-016-1087-y
CrossRef - Brito MA de. Pharmacokinetic study with computational tools in the medicinal chemistry course. Brazilian J Pharm Sci. 2011;47(4):797-805. doi:10.1590/S1984-82502011000400017
CrossRef - Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615-2623. doi:10.1021/jm020017n.
CrossRef

