
Manuscript accepted on :30-03-2021
Published online on: 30-03-2021
Plagiarism Check: Yes
Reviewed by: Dr. Avtanski, Dimitar B
Second Review by: Dr. Mustafa
Final Approval by: Dr. Ayush Dogra
Erni H. Purwaningsih1 , Anton Oertl2,3, Seruni K.U. Freisleben3,4 and Hans-Joachim Freisleben1,2*
, Anton Oertl2,3, Seruni K.U. Freisleben3,4 and Hans-Joachim Freisleben1,2*
1Department of Medicine, University of Indonesia, Jakarta-Depok, Indonesia
2Department of Medicine, Goethe-University Frankfurt/Main, Germany
3Asklepios Clinical Hospital and MVZ, Wiesbaden, Germany
4Department of Natural Sciences, University of Indonesia, Jakarta-Depok, Indonesia.
Corresponding Author E-mail: hj.freisleben@t-online.de
DOI : https://dx.doi.org/10.13005/bpj/2097
Abstract
Immune-suppressive agents such as methylprednisolone and cyclosporine exert tremendous side effects, because of high dosage and long-term application required for immune suppression after organ transplantation. Major side effects of methylprednisolone include bleeding of the gastro-intestinal tract, hypertension, and osteoporosis, whereas cyclosporine is nephrotoxic. Liposomes are phospholipid particles that allow delivery of drugs preferentially to the reticuloendothelial system. They can be prepared from phospholipids such as lecithin from soybean or egg yolk, other specific or modified lipids or from membrane-spanning tetraether lipid (TEL), which can be extracted and purified from archaeal cell membranes. One advantage in the use of liposomal application is reduced toxicity of many drugs. We report on various liposomal preparations of cyclosporine, methylprednisolone (L-MPL) and its palmitate derivative (L-MPLP). It has been documented that liposomal cyclosporine A (L-CsA), 1.75 mg/kg/day for seven days has potential for use as an immune-suppressive agent in rats with increased efficacy and decreased nephrotoxicity compared to commercially available forms of intravenous CsA. Liposomal methylprednisolone (L-MPL) 2 mg/kg, intravenously (IV), twice a week shows significantly prolonged cardiac allograft survival in rats and tissue-selective sequestration of the drug in comparison with the same dosage regimen of methylprednisolone in solution, administered daily. We report on organ distribution of L-MPLP in rats after intraperitoneal (IP) administration. Conclusion: Liposomal preparations of immunosuppressants have significantly higher immune-suppressive potential and lower toxicity than non-liposomal preparations. Bipolar TEL can be extracted, fractionated and purified from archaea to form stable liposomes which are extremely resistant, even to gastric fluid. Hence, TEL liposomes allow (besides IV and IP) for oral administration of immuno suppressants after organ transplantation with pharmacological and toxicological advantages as common liposomal phospholipid bilayer preparations.
Keywords
Absorption; Allograft; Cyclosporine; Gastrointestinal Stability; Immuno Suppressant; Liposomes; Methylprednisolone; Organ Transplantation; Oral Administration; Toxicity
Download this article as:| Copy the following to cite this article: Purwaningsih E. H, Oertl A, Freisleben S. K. U, Freisleben H. J. How Can Immuno suppression After Organ Transplantation be Made More Effective and Safer? – A Review on Liposomal Formulations with Consideration of Archaeal Tetraetherlipid. Biomed Pharmacol J 2021;14(1) |
| Copy the following to cite this URL: Purwaningsih E. H, Oertl A, Freisleben S. K. U, Freisleben H. J. How Can Immuno suppression After Organ Transplantation be Made More Effective and Safer? – A Review on Liposomal Formulations with Consideration of Archaeal Tetraetherlipid. Biomed Pharmacol J 2021;14(1). Available from: https://bit.ly/3rVnZkV |
Introduction
Organ transplantations are among the major health problems worldwide and especially in developing countries. The incidence of the organ rejection after transplantation is still high. Applying ineffective doses of immuno suppressive agents may be part of the problem Immuno suppressive drugs such as methylprednisolone (MPL) and cyclosporine A (CsA) exert tremendous side effects, especially because of high dosage and long-term application required for immuno suppression after organ transplantation. During the episodes of acute rejection, 500-1000 mg of MPL are given intravenously for several days. Because of the high dosage of glucocorticoids required for immuno suppression, side effects are common, such as bleeding of the GI tract, hypertension, and osteoporosis.1-3 Moreover, long-term medication with MPL will disturb the endogenous corticosteroid regulation. Complications related to systemic immuno suppression are major causes of morbidity and mortality following organ transplantation. The major unwanted side effect of high dose and long-term application of CsA is nephrotoxicity.1-3
Incorporation of therapeutic compounds into liposomes depends on the physicochemical properties of both the compound and the lipid. The most effective way for the incorporation of sufficiently lipophilic compounds is to mix them with the lipids from the beginning of preparation, so that they are preferentially inserted into the liposomal membrane via hydrophobic interaction. Normally, therapeutic concentrations do not require amounts of a respective compound too high to be taken up into the liposomal membrane by approximately 100% with this method. This was also shown for methylprednisolone and cyclosporine; however, liposomes prepared without special stabilizers lack stability and exert rapid loss of incorporated compound(s). Hence, a special aspect of this review is the stabilization of liposomes with incorporated immuno suppressants by means of archaeal tetraether lipid. This part will be described in detail.
Cyclosporine
Cyclosporine A (CsA) is a cyclic peptide found in Tolypocladium inflatum, Gams. It is an extremely hydrophobic molecule. As an immune-suppressive agent, CsA has a highly selective ability to inhibit the activation of T-cells. Although its site of action has not been defined precisely, it inhibits an early cellular response to antigenic and regulatory stimuli, primarily in populations of Helper-T-cells. Cyclosporine also causes a general reduction in the production and release of lymphokines in response to an antigenic stimulus. At higher concentrations, CsA inhibits expression of receptors for IL-2. Although CsA can inhibit the activation of Helper-T-cells, it does not prevent the stimulation of their clonal expansion by IL-2. It is potentially significant that CsA allows the expression of suppressor cell activity at concentrations that inhibit the induction of cytotoxic T-cells.4
Liposomal Cyclosporine A
The first reports on liposomal CsA (L-CSA) go back to the eighties of last century.5 In general, natural compositions of lecithin from soy beans or egg yolk were used, as well as defined lipid composition.6-8 Liposomal CsA was reported less nephrotoxic.9 In 1989, Vadiei et al.10 compared two liposomal CsA formulations of dimyristoyl phosphatidylcholine (DMPC): stearylamine (SA) (7:1 molar ratio) and DMPC: dimyristoyl phosphatidylglycerol (DMPG) (4:1 molar ratio) for IV application, at optimal drug:lipid ratio of 1:20 (w/w) for both formulations. According to their evaluation, DMPC:SA is advantageous over the second formulation.10
Later on, liposomes for CsA delivery have been prepared from more complex composition, phosphatidylserine / phosphatidylcholine, cholesterol sulfate, and lyso-phosphatidylcholine in a molar ratio of 3:4:2.11 The authors reported higher efficacy and lower nephrotoxicity. Further characterization and pharmacokinetic studies including oral administration and intermembrane exchange were conducted.12-17
The interaction and insertion of cyclosporine into biological and model membranes has widely been investigated and discussed.18-21 Comparing the structure of cyclosporine, a lipophilic cyclic peptide with 11 amino acids with similar compounds that were investigated in bilayer and tetraether lipid (TEL) membranes,22-24 valinomycin, a lipophilic cyclic peptide with 12 amino acids resembles cyclosporine most; however, alamethicin also exerts similar behavior, especially considering the interaction with cholesterol. Cyclosporine A interfered both with lateral lipid organization19 and probably with cholesterol binding sites, because addition of cholesterol to liposomal membranes reduced their binding capacity from the maximum of one CsA molecule per 19 lipid molecules.21 Although inserted deeply into the hydrophobic moiety of membranes, preferentially at the interface between fluid-crystalline and gel-analogous lipid domains, the single hydroxyl group of CsA may be oriented to and interact with the polar head groups of lipid molecules20 and thus, with cholesterol binding sites.25
Liposomal Preparations for Oral Administration
Liposomal preparations for oral cyclosporine delivery were investigated by several working groups.26-29 Modified liposomes for the oral delivery of CsA have been tested in vitro and in rats.30 Chitosan-modified liposomes (CS-Lip) were compared with Pluronic® F127-modified liposomes (PF127-Lip). Liposomes were prepared in a similar way as described above, finally extruded 6 times through polycarbonate filters of 200 nm pore size by means of a high-pressure homogenizer. 31-32 The composition was CsA, egg yolk phosphatidyl choline (EPC), and cholesterol at a 1:28:5 molar ratio. Pluronic® F127 (PF127), a non-ionic chemical polymer and cationic polysaccharide chitosan (CS), at a deacetylation degree of 94%, were added individually to yield hydrophilic non-ionic (PF127-Lip) and cationic (CS-Lip) polymer-modified liposomes. 30,33 The latter are known as mucoadhesive and the former as mucus-penetrating particles,34 which both had been shown in animals to increase the intestinal absorption of loaded drugs versus the absorption of free drug and from unmodified liposomes.
Size stability of these particles was tested in artificially simulated gastric fluid (SGF, pH 1.2) for 2 hours and simulated intestinal fluid (SIF, pH 6.8) for 6 hours. Initial diameters were 165.25±9.28 nm of unmodified liposomes (Lip), 172.82±15.79 nm of PF127-Lip, and 207.81 ±12.21 nm of CS-Lip. In SGF, only PF127-Lip remained stable, where as Lip significantly increased and CS-Lip significantly decreased in size. In SIF, Lip decreased in size, whereas PF127- Lip increased. Although significant these changes were still moderate within a range of less than 50 nm. On the contrast, CS-Lip changed tremendously in size, after 2 hours large aggregates precipitated and 40% of the liposome-loaded drug was released into the supernatant.30 On the other hand, release profiles of all three preparations in ethanol-containing PBS within 24 hours were similar without burst phenomena indicating that CsA was well incorporated into the liposomes and not essentially influenced by the modifications. Entrapment efficiency was 85% in Lip, 82% in CS-Lip, and 90% in PF127-Lip, and release from all preparations was up to 85% of the incorporated CsA within 24 hours.30
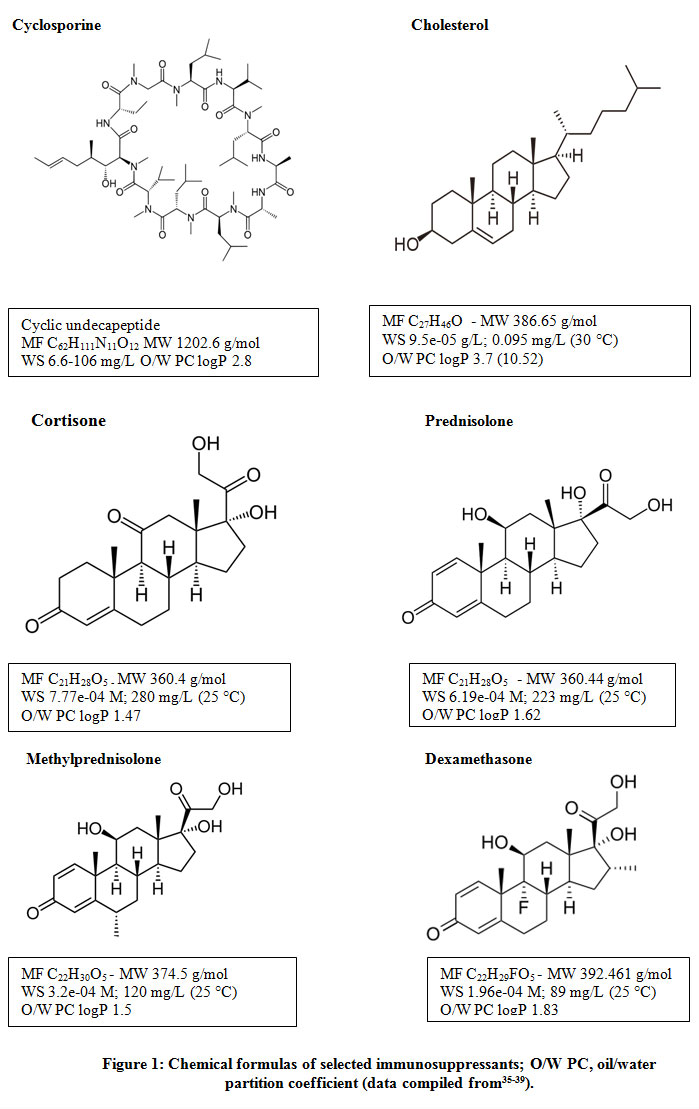 |
Figure 1: Chemical formulas of selected immunosuppressants; O/W PC, oil/water partition coefficient (data compiled from35-39). |
For the determination of the penetration into intestinal mucus and tissue in rats coumarin-6 was incorporated into the liposomes as hydrophobic model substance which allows for fluorophotometric measurement. Fluorescence was measured in the duodenum, jejunum and ileum and was highest in the mucus with CS-Lip and highest in the tissue with PF127-Lip. In other words, CS-Lip get stuck in the mucus, typical of mucoadhesive particles and don’t reach enterocytes in intestinal tissue, which means that they are not suitable for effective oral drug delivery. In contrast, PF127-Lip are able to effectively deliver their drug load into the intestinal tissue.30
Pharmacological Parameters of Csa And L-Csa
Oral bio availability of CsA varies from 20 to 50 %. About 60-70 % of the drug in whole blood is contained in erythrocytes, 10-20 % contained in leukocytes. Cyclosporine has a plasma half-life of 5.6±2 hours and a plasma clearance of 5.9±1.0 mL/min/kg. The volume of distribution is 1.2±0.2 L/kg. Oral treatment with a dose of 15 mg/kg BW is initiated 4 to 24 hours prior to transplantation, once daily and continued for one to two weeks after transplantation. Thereafter, the dosage is reduced each week until a maintenance dose of 3 to 10 mg/kg/day is reached. Dosage is generally guided by signs of renal toxicity, as judged from changes in creatinine clearance.4
In the study of Chen et al., intestinal absorption and bio availability of liposomal CsA after oral administration of 10mg/kg BW were determined by the measurement of Cmax [µg/mL], Tmax [h], and AUC0→t [µg.h/mL] in the blood of Sprague-Dawley rats. The values of CsA in plasma differed between PF127-Lip and CS-Lip roughly by a factor of two; Cmax (1.37 ± 0.15 vs. 0.79 ± 0.10) and AUC0→t (11.59 ± 0.7 vs 6.30 ± 097) were significantly higher and Tmax (1.68 ± 0.29 vs. 3.67 ± 0.58) significantly lower in PF127-Lip than in CS- Lip; the values with unmodified liposomes were in between.30
Cyclosporine encapsulation using glyceryl monooleate-poloxamer 407 nano particles (poloxamer 407 is identical with Pluronic® F127) yielded 85% encapsulation efficacy and higher relative oral bio availability in beagle dogs of about 178% versus Sandimmun Neoral as reference.40 The relative oral bio availability of L-CsA encapsulated in soy phosphatidylcholine (SPC) liposomes containing sodium deoxycholate instead of cholesterol was slightly more than 120% as compared with cholesterol-containing SPC liposomes or Sandimmun Neoral.31
If CsA cannot be administered orally, it must be infused slowly over a period of 2 to 6 hours or longer. The daily dose should be only one-third the oral dose (usually 5-6 mg/kg BW).4 Two hours after IV injection of L-CsA in rats at a single dose of 2 mg/kg, the distribution was increased two-fold in liver and spleen tissues compared to CsA in saline. Intravenous administration of 10 mg/kg CsA in saline was demonstrated to exhibit exaggerated nephrotoxicity in renal ischemia induced by contralateral nephrectomy in rats, whereas serum creatinine in animals, which had received liposomal CsA, returned to control levels within 96 hours. In liver transplant models in rats, the dose of 1.75 mg/kg of L-CsA resulted in significantly prolonged survival rates for about 93 days as compared to the group, which had received the same dose of CsA in saline.11
Dexamethasone
Dexamethasone is a glucocorticoid, which has the capacity to suppress immune responses. Although considered to be immune suppressive, therapeutic doses of glucocorticoids do not significantly decrease the concentration of antibodies in the circulation. The immune response is initiated by the interaction of an antigen with macrophages and with antibodies on the surface of B-lymphocytes. Glucocorticoids interfere with the function of macrophages in several ways: firstly, they inhibit the action of MIF (migration-inhibitory factors), thereby promoting the emigration of macrophages from affected areas; secondly, they inhibit the processing and display of antigens by interfering with the facilitating actions of γ-interferon; thirdly, they inhibit the synthesis and release of IL-1. More importantly, IL-1 participates in the activation of resting T-lymphocytes when they come in contact with processed and histocompatibility antigens displayed on the surface of activated macrophages. Hence, glucocorticoids suppress the activation of T-cells by several mechanisms. Recently, dexamethasone has come into the focus with SARS-CoV2 infections and COVID-19,41 but this is not the topic to be discussed here.
Liposomal Dexamethasone
Tanaguchi et al.42 compared the incorporation rates of dexamethasone (DM), dexamethasone acetate (DA), dexamethasone valerate (DV), and dexamethasone palmitate (DP) into liposomes at a ratio of 16 mM lipid and 0.5 mM steroid (or its respective derivative), which is equivalent to 3.125 mol% steroid. They determined the water solubility in physiological buffer pH 7.4 at 37oC as 80.5, 29.6, 1.3, and 0.052 mg/L, respectively. Chloroform/water partition coefficients (PC) were 9, 657, 8780, and not measurable for DP which values are comparable to DM logP 0.95, DA logP 2.8, DV logP 3.9 and a higher, not measurable, value for DP.42
The authors used sonication and subsequent filtration through a polycarbonate filter with one µm pore width. Incorporation rates were increasing (slightly depending on sonication time between 2.5 and 20 min), 90.5-92.0 % (DM), 94.7-96.0 % (DA), 99.5-99.6 % (DV), and 100% for DP. Various lipid compositions comprised egg yolk lecithin (EYL), dioleoyl-phosphatidylcholine (DOPC), stearylamine (SA), diacetyl-phosphate (DCP) with similar incorporation rates; only dipalmitoyl-phosphatidylcholine (DPPC) had lower DM incorporation rates of 78.5%.42
The authors also investigated the influence of cholesterol on the incorporation of steroids (DM and derivatives): cholesterol up to 4 mM concentration increased free steroid in the preparations concentration-dependently from 20 to 40 mg/L (DM); from slightly above 10 to 20 mg/L (DA); much less with DV (below 3.5 mg/L); and not at all with DP (0 mg/L).42 It was concluded that steroids interact differently from DP with lipids in the membrane and that cholesterol competes with them.43 Steroids may be oriented horizontally, so that the hydroxyls remain hydrated, whereas DP may be inserted into the membrane perpendicularly via its palmitate side chain, similar to cortisone-21-palmitate.44,45
Prednisolone and Methylprednisolone
Liposomal Prednisolone
Prednisone and Prednisolone are mainly used for the treatment of rheumatic and atherosclerotic diseases. Liposomal preparations have been reported with prednisolone,46-48 but liposomal preparations have not been applied for immuno suppression and are therefore not discussed here.
Liposomal Methylprednisolone (L-MPL)
Methylprednisolone is a glucocorticoid with immuno suppressive activity similar to what was already described for dexamethasone. Mishina et al. considered the optimum formulation being EPC and phosphatidylglycerol (PG) at molar ratio of 9:1 and 5 mol% MPL.49 The authors refer to50-52 and consider 10% of negatively charged lipid in the EPC membrane best compromise for incorporation of MPL into and its retention in liposomal membranes. Liposomes were stable overnight, but lost 70% of MPL within one week.49
Therefore, it was decided to stabilize L-MPL with archaeal tetraether lipid. Liposomal MPL was prepared from EPC (= EYL) and phosphatidylglycerol (9:1 molar ratio) with 5 mol% of MPL.53,54 Ethanolic solutions of these mixtures (20mg/mL) were evaporated under reduced pressure (250 mbar) in a round-bottom flask at 40oC of the water bath of a Büchi Rotavapor. The dry lipid film was stored at RT for 12 hours in a vacuum desiccator in order to remove residual moisture. Subsequently, the lipid film was suspended in PBS, pH 7.4, at an amount to result in 15-20 mg of lipid mixture per mL of buffer. This suspension was shaken by hand with two glass beads added into the flask. The resulting suspension of large multilamellar vesicles (LMV) was sonicated and extruded (5 to 7 times) through Liposofast® polycarbonate filters (pore size 100 nm).55
To separate free (not encapsulated) drug, the liposomal suspension was applied to an open Sephadex G-75 chromatography column. All liposomal preparations were routinely checked in a Malvern Particle Sizer and several representative preparations in addition by electron microscopy.24 The polycarbonate filter extrusion resulted in unilamellar vesicles of roughly 100 nm in diameter, different preparations varied from above 50 to 120 nm, but each single preparation had to be uniformly size-distributed in order to be used for further investigation.
For the incorporation of MPL into liposomes, phosphatidylglycerol (PG) was admixed to EPC at 10 mol% or (instead of PG) TEL extracted and purified from Thermoplasma acidophilum (T.a.) at 2.5 or 10 mol%; in addition, pure TEL liposomes (100 mol%) were used. In all cases, 5 mol% MPL were applied for liposomal incorporation resulting in molar liposomal ratios of 9.4-9.5:0.5-0.6 (lipid:MPL), equivalent to incorporation ratio of about 100% in all cases and 0.5-0.7 mg MPL per mL of liposomal suspension. Since liposomes prepared according to this method lack stability (they must be freshly prepared for each test series), Oertl et al.56,57 and Bräutigam et al.58,59 suggested stabilization of L-MPL preparations with TEL according to Freisleben et al.60 (Table 1).
Table 1: Stability of liposomal MPL preparations.
| Lipid composition | Molar ratio | Negatively charged lipid [mol %] | Stability: Loss of MPL within one week |
| EPC/PG | 9:1 | 10 | 65-75% |
| EPC/TEL | 9:1 | 10 | 14-17% |
| EPC/TEL | 39:1 | 2.5 | 18-20% |
| TEL | – | 100 | 20-23% |
Footnote: PG, phosphatidylglycerol; EPC, egg phosphatidylcholine; TEL, tetraether lipid.
Animal Experiments and Pharmacological Parameters.
The steroid MPL is effective when given orally; its bio availability is 82 ± 13 %. In the plasma, 90% or more are reversibly bound to proteins (albumin and globulin) under normal conditions. MPL clearance is 6.2 ± 0.9 mL/min/kg.4 MPL has a short plasma half-life, but the liposomal formulation L-MPL markedly prolonged plasma circulation time and led to sequestration of the steroid into the lymphatic tissues. Terminal half-life was dramatically extended from 1.5-2.5 hours for MPL to more than 30 hours for L-MPL (Table 2).49 The distribution volume of L-MPL 1-2 hours after intravenous injection of a 2 mg/kg bolus in rats was significantly increased from 1.2 ± 0.2 L/kg of MPL in lymphatic tissues: in spleen 77-fold; in thymus 27-fold and in liver 9-fold. In liver and spleen MPL remained detectable for 26 days after L-MPL injection confirming the tissue-selective sequestration of the drug.49
Table 2: Pharmacokinetic parameters of MPL and L-MPL (data compiled from49).
| Parameter | MPL | L-MPL | Unit | Remarks |
| AUC | 339 | 1093 | ng x h/mL | area under the curve |
| Clearance (CL) | 5.04 | 1.83 | L/h/kg | apparent systemic clearance |
| CLD | 2.24 | 67.08 | L/h/kg | distribution clearance |
| t1/2 | 0.48 | 30.13 | h | terminal half-life |
| MRT | 0.42 | 11.95 | h | mean residence time |
| MRTt | 0.14 | 11.73 | h | mean residence time in tissues |
| Vc | 1.39 | 0.39 | L/kg | central volume of distribution |
|
Vss |
2.1 | 21.87 | L/kg |
total volume of distribution |
Male Lewis RT1 rats served as recipients of cardiac allografts from Lewis x Brown Norway F1 (LEWxBN)F1 hybrids.53,54,56,57,61 Hearts were anastomosed to the abdominal great vessels using standard microvascular techniques. Ventricular contractions were assessed daily by palpation, and rejection was defined as the day of cessation of heartbeat.53,56,57,59,61
The dosage of L-MPL of 2 mg/kg BW, intravenously twice a week led to cardiac allograft survival of 20.8 ± 6.5 days (maximum 30 days), whereas with the same dose of non- liposomal MPL (from a conventional commercial preparation) the allograft was rejected after 7.8 ± 1.0 days, similar to the untreated control group (9.2 ± 1.2 days). The dosage of 4 mg/kg BW, intravenously once a week, also significantly prolonged cardiac allograft survival in rats to 17.5 ± 2.8 days, which was comparable with daily administration of 2 mg/kg BW of MPL in solution (17.0 ± 2.7 days). Only the daily injection of 4 mg/kg BW of non-liposomal MPL (i.e., seven times higher dosage) led to results comparable to the treatment with liposomal MPL.53,61
Bilaterally nephrectomized LEW rats were recipients of BN kidney transplantation (Tx).54,58,59 L-MPL was administered IV, 2 mg/kg BW, twice a week or 4 mg/kg BW, once a week. Distribution of L-MPL was determined in liver, spleen and thymus between one and two hours after injection.49 Renal allograft survival with 4 mg/kg BW of L-MPL (IV once a week) significantly prolonged allograft survival to 20.2 ± 7.4 days (comparable with daily administration of 4 mg/kg MPL in solution: 19.0 ± 6.8 days) vs. acute rejection of 8.5 ± 0.5 days (p < 0.001). MPL + empty liposomes injected separately once a week was ineffective (8.8 ± 0.5 days).54
Liposomal Methylprednisolone Palmitate (L-MPLP)
Methylprednisolone-palmitate (MPL-P) is a prodrug synthetized for the same reason as the derivates of dexame-thasone (DA, DV, DP), in order to increase and stabilize their incorporation into liposomal membranes; in case of EPC alone MPL-P was incorporated at a maximum of 79%. The addition of 2.5 mol % of TEL from Sulfolobus acidocaldarius (TEL- S.a.) increased the incorporation rate to approximately 95% (range 94-97%) at concentrations of 4-10 mol% added to the original suspension. TEL itself was incorporated at a rate of 85-86% (Table 3).62 In detail, the incorporation of 2.5 mol% TEL into EPC at various concentrations yielded in: EPC concentration 0.52 mM, TEL 0.011 mM, incorporation rate 84.6%; EPC concentration 2.03 mM, TEL 0.044 mM, incorporation rate 86.7%; EPC concentration 2.54 mM, TEL 0.054 mM, incorporation rate 85.0%.
Table 3: Incorporation of TEL and MPLP into EPC liposomes (data compiled from62)
| Input MPLP | 4 mol% | 5 mol% | 10 mol% |
| Input
EPC 1.7 mM |
Detected in EPC liposomes | ||
| 1.09:0.028 mM* | 0.86:0.034 mM* | 0.73:0.013 mM* | |
| Percentage of incorporation | MPLP
64.2% |
MPLP
79.0% |
MPLP
17.8% |
| Detected in EPC liposomes stabilized by 2.5 mol% TEL-S.a. | |||
| Input
EPC:TEL 1.7:0.04 mM |
mM concentrations* EPC:TEL:MPLP 1.02:0.022:0.039 | mM concentrations* EPC:TEL:MPLP 1.27:0.027:0.066 | mM concentrations* EPC:TEL:MPLP 0.52:0.011:0.050 |
| Percentage of incorporation | TEL 86.7% MPLP 94.1% |
TEL 85.0% MPLP 97.1% |
TEL 84.6% MPLP 95.2% |
Footnote: EPC, egg yolk phosphatidylcholine, *measured by enzymatic reaction photometrically at wavelength λ = 490 nm; MPLP = methylprednisolone palmitate; TEL = tetraether lipid *measured from spots on TLC with scanner detection.
Liposome suspensions were formed with input concentrations of 1.7 mM or 3.4 mM EPC, 2.5 mol% TEL, and 2-10 mol% MPLP. Best results were obtained with 4-5 mol% of MPLP. In liposome suspensions containing MPLP in 1.7 mM EPC, the highest amounts of EPC and MPLP were found in 1.0 mL fractions 2-4 after Sephadex G-75 column gel filtration chromatography (GFC) with MPLP distribution values in fraction 3 around 40-43% (43.1% at MPLP 4 mol% and 40.0% at MPLP 5 mol%) and in liposomal mixtures containing TEL, distribution value in fraction 3 was 47% at 4-5 mol% MPLP input62 (for details see Materials and Methods section).
Particle size stability of the liposomes was determined; L-MPLP is stable at 20°C for 21 days, although the size tends to become larger and size distribution broadens. EPC-MPLP liposomes are less stable (Table 4, data collected from63,64).
Table 4: Stability of liposomes consisting of EPC, TEL (2.5 mol%) and MPLP (5 mol%).
| Temperature
20º C |
Days of storage | |||
| 0 | 3 | 9 | 21 | |
| EPC:TEL:MPLP | 111 nm | 132 nm | 146 nm | 162 nm |
| EPC:MPLP | 91 nm | 113 nm | 185 nm | aggregation |
Pharmacological Parameters
Organ distribution of MPL/MPLP was measured as µg/g tissue after IP injection of L-MPLP (data collected from65).
Table 5: MPL/MPLP in organs [µg/g tissue].
| Minutes | Liver | Thymus | Spleen | Bone marrow | Kidney left | Kidney right |
| 30 | 10.66 | 1.23 | 3.28 | 0.65 | 0.81 | 3.25 |
| 60 | 13.48 | 5.35 | 5.88 | 1.01 | 3.49 | 6.71 |
| 210 | 16.33 | 8.22 | 2.52 | 2.70 | 1.40 | 5.47 |
| average | 13.54 | 4.02 | 3.48 | 2.23 | 2.15 | 5.16 |
Footnote: MPL = methylprednisolone; MPLP = methylprednisolone palmitate; L-MPLP = liposomal methylprednisolone palmitate.
Table 6: MPLP in liver [µg/g tissue] after IP injection (data from65).
| Minutes | Liposome | MPL | MPLP | L-MPLP |
| 30 | 0.28 | 0.30 | 0.01 | 0.88 |
| 60 | 0.24 | 0.08 | 0.50 | 0.84 |
| 90 | 0.27 | 0.21 | 0.20 | 1.60 |
| 150 | 0.30 | 0.32 | 0.44 | 1.39 |
Footnote: MPL = methylprednisolone; MPLP = methylprednisolone palmitate; L-MPLP = liposomal methylp

