
Manuscript accepted on :31-Oct-2020
Published online on: --
Plagiarism Check: Yes
Reviewed by: Flavio Palmieri
Second Review by: Ahmed salah Naser
Final Approval by: Ian James Martin
Leana Rich M. Herrera
Department of Physical Sciences, College of Science, Polytechnic University of the Philippines, Manila City, Philippines
Corresponding Author Email: lrmherrera@pup.edu.ph
DOI : https://dx.doi.org/10.13005/bpj/2060
Abstract
The rapid transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has resulted to the death of hundreds of thousands of people worldwide. With the devastating effects on the economy and healthcare system of many countries, it is crucial to acceleratevaccine development against SARS-CoV-2. Thus, thisworkutilizedimmunoinformaticsto efficiently design a novel multi-epitope vaccine that can potentially induce immune response through the immunogenic, and abundantly expressed structural proteins in SARS-CoV-2. Epitopes were screened and evaluated using various immunoinformatics tools and databases. Antigenicity, allergenicity, and population coverage were assessed. Epitopes were adjoined to form a single vaccine construct (Covax),linked with 50S ribosomal protein as an adjuvant. Physicochemical properties, cross-reactivity, antigenicity,andallergenicityof Covax were evaluated. The tertiary structure of Covax was modeled, refined and validated for docking with toll-like receptor 4 (TLR4). Binding affinity of Covax-TLR4 was estimated and compared with TLR4-adjuvant as control. Lastly,the immune response with Covax was simulated and compared withadjuvant alone. Total of 33 epitopes from S (21), E (3), M (5),and N (4)proteins were merged in Covax. These include epitopes on thereceptor-binding motif (RBM) of S protein known to beessential in the viral attachment. In silico evaluations classified Covax as stable, antigenic, and non-allergenic. Epitopes were estimated to have large worldwide population coverage, especially in areas with high infection rates, indicating broad potential efficacy of Covax as a vaccine for the most affected populations.Results in this work showed that Covax can bind to TLR4 whichindicates potential immunogenicity and superior properties necessary for a successful vaccine. Overall, this work efficiently minimized time, effort and cost in designing a candidate vaccine against SARS-CoV-2. In vitro and in vivo studies on Covax are anticipated.
Keywords
Epitopes; Immunoinformatics; In Silico; Receptor-Binding Motif; SARS-Cov-2; Vaccine
Download this article as:| Copy the following to cite this article: Herrera L. R. M. Immunoinformatics Approach in Designing a Novel Vaccine Using Epitopes from All the Structural Proteins of SARS-CoV-2. Biomed Pharmacol J 2020;13(4). |
| Copy the following to cite this URL: Herrera L. R. M. Immunoinformatics Approach in Designing a Novel Vaccine Using Epitopes from All the Structural Proteins of SARS-CoV-2. Biomed Pharmacol J 2020;13(4). Available from: https://bit.ly/3lJn2Jq |
Introduction
The coronavirus disease 2019 (COVID-19)pandemicis caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) outbreak which was first identified in Wuhan, China in December 2019 (Li Q et al., 2020). This novel virusattacks vital organs,mainly the lungswhich canprogress into fatalpneumonia and acute respiratory distress syndrome (Wang et al., 2020). SARS-CoV-2 can be easily transmitted throughdirect contact, fomites, and respiratory droplets (Galbadage, et al., 2020). However, infected individuals may or maynot have symptoms which commonly include fever, cough, and fatigue (Huang et al., 2020).Since SARS-CoV-2 has just been discovered recently, very little is known about its pathogenesis. Further investigative studies on the nature of this novel virus must be conducted.
Currently known human coronaviruses (HCoV) including HCoV-229E andHCoV-NL63belong to genus Alphacoronavirus while HCoV-OC43, HCoV-HKU1, Middle East Respiratory Syndrome (MERS), SARS-CoV and SARS-CoV-2 belong to genusBetacoronavirus, family Coronaviridae(Al-Tawfiq et al., 2014). Phylogenetic studies showed SARS-CoV-2shared 96% and 79.6% sequence identity with bat coronavirus and SARS-CoV, respectively (Zhou et al., 2020).Coronaviruses are enveloped viruses containingpositive sense single-stranded RNA as genetic material. It has approximately 26-30kbencoding non-structural proteins (NSP) and structural proteins including spike (S), envelope (E), membrane (M) and nucleocapsid (N) (Ahmed SF, et al., 2020).Structural proteins in coronaviruses have different roles yet with integrative functions. Glycosylated S protein is consist of two subunits S1 and S2forming a trimeric structure on the surface of the virus similar to spikes of a crown (Walls et al., 2016). S protein is vital in cell host attachment, fusion, and entry. It contains a receptor-binding motif (RBM) which binds with angiotensin-converting enzyme2 (ACE2)receptor of the host cell facilitating its entry (Hoffmann et al., 2020). These crucial roles make S protein the most valuable target in vaccines and therapeutics (Zhu et al., 2013).Coronavirus E protein is a single-pass transmembrane protein which self-assembles in host membrane acting as a viroporinto allow the transport of ions (Verdia-Baguena et al., 2013; Wilson et al., 2004). It is the smallest structural protein in corona viruses yet plays important roles in assembly, viral release, and contributes in the virulence of HCoVs (Nieto-Torres et al., 2014). N protein is the only structural protein known to bind and encapsidate the genomic RNA (gRNA) of corona viruses (Ma et al., 2010; Kuo et al., 2016). Itis also abundantly expressed in the host cell during infection making it an excellent target in constructing vaccines to induce activation of cytotoxic (CTL) and helper T lymphocytes (HTL). This multi-functional protein plays important roles in organization of viral particles through its association with gRNA and with M protein (McBride et al., 2014). M proteinis a small transmembrane protein integrated in the membrane of all corona viruses. Its participation for intra cellular viral assembly is S protein-dependent. As the most abundant structural protein in corona viruses, M protein is also involved in packaging gRNA, regulation of replication, and morphogenesis (Neuman BW, et al., 2010; Hu Y, et al., 2003). A recent study showed that T-cell responses elicited by SARS-CoVstructural proteins are more immunogenic than the nonstructural proteins (Li et al., 2008). Since the genetic sequence of SARS-CoV-2sharesa close identity with SARS-CoV, generating epitopes from the sequences of these structural proteins is a good strategyto developan immunotherapeutic agent against COVID-19.
SARS-CoV-2 pandemic warrants urgentdiscovery of anti-viral drugs and vaccines. Immunoinformatics and data mining have efficiently hastened and reduced the cost required in experimental immunology to identifyvaccine candidates and biomarkers (Vaishnav et al., 2015; Oyarzún et al., 2016). Just to mention a few, immunoinformatics has aided in designing vaccines that demonstratedsafety, and promising resultsagainst herpes simplex virus type 2and human influenza H1N1, both of which are currently under preclinical and clinical trials, respectively(Pan et al., 2012; Pleguezuelos et al., 2020). Thus, the primary goal of this work is to apply immunoinformatics tools, and databases to efficiently design a vaccine that can potentially induce immune response against SARS-CoV-2. Several vaccine candidates against SARS-CoV-2are already in clinical trials; however, these cannot guarantee success—the pursuit for a safe and effective vaccine is far from being resolved. This work focused in designing a multi-epitope vaccine from all the structural proteins of SARS-CoV-2.Constructing a multi-epitope vaccine could be more advantageous than single peptides. These include larger population coverage, simultaneous induction of immune response due to the adjoined promiscuous epitopes, linkage of an adjuvant which increases immunogenicity of the epitopes, and exclusion of sequences that can result to adverse effects.
Methodology
Retrieval of Sequences
Thirty three SARS-CoV-2isolates from different areas around the world including Taiwan, Turkey, Sweden, Iran, Brazil, Australia, Bangladesh, Columbia, Czechoslovakia, France, Greece, India, Peru, Serbia, South Africa, Spain, China, Germany, Guam, Hong Kong, Israel, Italy, Kazakhstan, Malaysia, Nepal, Pakistan, Puerto Rico, Korea, Sri Lanka, Thailand, California USA, Uruguay, and Vietnamwere retrieved inthe NCBI Virus database (date retrieved: June 1, 2020).Only those with complete sequences were retrieved, and environmental sources were excluded. The S, E, M, and N protein sequences from these isolates which are accessible in supplementary file 1 (Supp1), were aligned per protein using Clustal Omega. To identify conserved fragments, protein variability analysis was conducted in Protein Variability Server using Shannon entropy threshold of 1.0 (Shannon 1948).Resulting conserved sequences were used for the screening of epitopes.In addition, the amino acid sequences of S, E, M, and N proteins from HCoV-229E, MERS, HCoV-HKU1, HCoV-OC43, SARS-CoV, and SARS-CoV-2 (NC_002645.1, NC_019843.3, NC_006577.2, NC_006213.1, NC_004718.3, and NC_045512.2) were also retrieved from the National Center for Biotechnology Information(NCBI)for cross-reactivity analysis.
Identification of Linear B Lymphocyte (BL) Epitopes
The positions of extravirion sequences forSARS-CoV-2S protein (13-1213), E protein (1-16), and M protein (2-19) are fully annotated in the UniProt database. These extravirion sequences were used to screen for linear B lymphocyte (BL) epitopes. N protein doesn’t have extravirion sequences; thus, it was not included in BL epitope screening.Default parameters in Emini Surface Accessibility (ESA), BepiPred Linear Epitope (BLE),Kolaskar&Tongaonkar Antigenicity (KTA) tools in Immune Epitope Database (IEDB), and ABCPredServer were used to generate the best linear BL epitopes. Surface accessibility is an important factor to consider in predicting potent epitopes. ESA scale estimates the accessibility of a hexapeptide sequence and calculates a score indicating its probability to be found on the surface (Emini et al., 1985). BLE prediction tool utilizes the combination of hidden Markov model and propensity scale method wherein the residues with scores above a certain threshold are presaged as epitopes (Jespersen et al., 2017). KTA predicts potential epitopes through amino acid residues and the frequency of their occurrence as epitopes based from experimental data. As applied to a large number of proteins, this tool was validated by Kolaskar and Tongaonkar (1990)to be 75% accurate, making it superior than other known methods in predicting epitopes. ABCPred uses artificial neural network and predicts BL epitopes with 65.93% accuracy (Saha&Raghava, 2006). Epitopes which overlapped with glycosylation and cleavage sites were excluded. Consensus BL epitopes with 12-15 residues from at least 2 servers were saved for further evaluations.
Identification of Helper T Lymphocyte (HTL) Epitopes
The complete amino acid sequence of S, E, M and N proteins fromSARS-CoV-2were used to screen for helper T lymphocyte (HTL) epitopes. HTL epitopes and their binding affinity with MHC II alleles were identifiedusingNetMHCIIpan-3.2 in IEDB server. This method was builton an extended data set of quantitative MHC–peptide binding affinity which has improved the performance of peptide binding prediction (Jensen KK, et al., 2018).HTL epitopes were obtained in this tool using the most frequent MHC II alleles (Greenbaum et al., 2011), and IC50 <500 nmol/dm3 which classifies epitopes as good binders (Jensen et al., 2018). Resulting epitopeswith 11 residues binding to almost allMHC II alleles from the list were saved for further evaluations. The list of the most frequent MHC II alleles used in the study can be accessed in supplementary file 2 (Supp2).
Identification of Cytotoxic T Lymphocyte (CTL) Epitopes
Completeamino acid sequence of S, E, M and N proteins fromSARS-CoV-2 were screened for epitopes binding with the most frequent MHC I alleles (Weiskopf et al., 2012) (Supp2).IEDB Proteasomal cleavage/transporter associated with antigen processing transport (TAP)/MHC class I combined predictor tool which employs NetMHCPan-2.0 method was utilized; thereby, consideringproteosomal processing, TAP transport, and binding affinity in predicting CTL epitopes altogether. NetMHCPan-2.0 is trained using the broadest set of available MHC I binding data which can be used for large out-bred populations (Hoof et al., 2009). Peptides with 9-11 residues, IC50 < 500 nmol/dm3, proteasome-processingscore> 1.0, TAP score > 1.0,and thosewhich covermost of the frequent MHC I alleles were saved for further evaluations.
Evaluation of Predicted Epitopes
Predictedepitopes were further evaluated for antigenicity, allergenicity, cross-reactivity, % conservancy, and cross-protection.Antigenic epitopes were identified usingthreshold ≥ 0.5 in Vaxijen 2.0 Server with a virus as a target. Vaxijen is an alignment-independent method which predicts antigen based on the physicochemical properties of proteins with 70% to 89% accuracy (Doytchinova& Flower, 2007). Potential allergenic sites were assessed using AllergenFP v 1.0. The highest Tanimoto similarity index of an epitope matching an allergen sequence from the database is used to determine if the epitope is a probable allergen (Dimitrov et al., 2014). To avoid potential autoimmune reactions, predicted epitopes were queried for possible hits against human protein sequences in UniProtKB and SwissProt databases using default parameters in protein-protein BLAST (BLASTp). Possible cross-protection offered by each epitope against 6 other HCoV strains was also investigated inIEDB Conservancy Analysis Tool (IEDB-CAT).Predicted epitopes which passed all these evaluationswere selectedfor inclusion in the vaccine.
Estimation of Population Coverage
The use of multi-epitopes could have larger population coverage (PC) for a vaccine. In this work, worldwide human PC were estimated using the Population Coverage tool in IEDB. The PC was calculated from each set of HTL and CTL epitopes as the vaccine will consist of multi-epitopes.In addition, PC in areas where infection rates of SARS-CoV-2are high (North America, Europe, East and Northeast Asia)were also estimated. This tool efficiently maximizes the population coverage of epitopes while minimizing the number of epitopes that must be included in a vaccine (Bui et al., 2006).
Construction of the Multi-Epitope Vaccine
An adjuvant and the selected BL, HTL, and CTL epitopes from S, E, M and N proteins of SARS-CoV-2were included in the vaccine—Covax. Overlapping CTL and HTL epitopes for the same protein were shortlisted using BL epitopes as template. For overlapping CTL and HTL epitopes which do not overlap with any BL epitopes, CTL epitopes were shortlisted using HTL epitopes as template. Within the same type of lymphocytes, overlapping epitopes for each protein were also merged as continuous peptides. To form the multi-epitope construct, BL and HTL epitopes were adjoined together using GPGPG linkers while CTL epitopes were adjoined using AAY linkers. For each type of lymphocyte, peptides from the same protein are arranged next to each other according to their positions in their respective proteins. For the adjuvant, sequence of 50S ribosomal protein (50S RP) from Mycobacterium tuberculosis strain ATCC 25618 / H37Rv (UniProt ID: P9WHE3) was linked with the N-terminus of the multi-epitope construct via flexible alpha-helix-forming EAAAK linker. In the complete Covaxconstruct, adjuvant is followed by each set of CTL, then HTL, and lastly BL peptides at the end to make it more accessible. Finally, Valine was added at the N-terminus of the vaccine construct to increase its half-life.
Evaluation of Physicochemical Properties, Antigenicity, Allergenicity, and Cross-Reactivity of Covax
The cross-reactivity of the candidate vaccine against proteins expressed in humans was investigated using default parameters in BLASTp. Models, non-redundant refseq proteins, and uncultured environmental samples were excluded. Allergenicity and antigenicity of the candidate vaccine were further assessedin AllergenFP v1.1 and Vaxijen 2.0 servers, respectively.Molecular weight, amino acid composition, isoelectric point (pI), half-life, extinction coefficient, instability index, thermostability (aliphatic index) and grand average hydropathicity (GRAVY) of Covax were calculated using ProtParam tool in ExPASy server.
Secondary Structure and Tertiary Structure Modelling
Percentage composition of secondary structures in Covaxwas estimated using GOR4 web tool. Intrinsically disordered regions were determined using GlobPlot2v2.3 server. Tertiary structures of Covaxand 50S RP were modelledusing usingGalaxyTBM tool. This tool employs multiple-template approach and ab initio modelling, evaluated to be one of the top TBM servers (Ko et al., 2012). Human toll-like receptor 4 (TLR4) crystal structure (PDB ID: 4G8A) was also retrieved. The ligands from the PDB structure were removed and only the monomeric form of TLR4 was used. The initial tertiary structure models of Covax, 50S RP, and retrieved crystal structure of human TLR4 were further refined in GalaxyRefine server. To check the validity of refined structures, ERRAT,Verify3D, and Rampage servers were employed. ERRAT analyzes the nonbonded atom to atom interactions of the input structure as compared with crystallography structures (Colovos&Yeates, 1993). Verify3D measures the compatibility of the tertiary structure model with its protein sequence which is then compared with the results of good structures (Eisenberg et al., 1997). Rampagegenerated Ramachandran plots showing the percentage of residues lying within the favoured, allowed, and disallowed regions. The best refined tertiary structure models were viewed inPymol and were used for protein-protein docking. Covax was further investigated for structural BL epitopes.
Identification of Structural B-Cell Epitopes
BL epitopesincorporated in Covaxmust be accessible and protruded enough soB-cell receptors (BCRs)can bind to it. Ellipro was utilized in the study to identify structural epitopes on the tertiary structure model of Covax. To date, it is the best structure-based algorithm amongst the others (Ponomorenko, et al., 2008). It predicts conformational and linear BL epitopes based from protrusion index (PI) of a residue, and provides PI score for each protruded sequence.
Molecular Docking Interaction of Covax-TLR4 and Peptide-HLA
An antigen must bind to specific immune receptorsto elicit an immune response. Thus, thiswork conducted data-driven docking to assess possible interactions between Covaxcontaining 50S RP as its adjuvant, withthe human TLR4 usingClusProserver.This toolperforms rigid docking based on Fast Fourier Transform by conducting conformation sampling, complex pair-wise root-mean-square deviation, and energy minimization step to yield docked complexes togetherwith their calculated binding energy scores (Kozakov et al., 2017). In this study, the best complex pose in terms of the lowest energy and dockingpositions was chosen and viewed in Pymol. The tertiary structure models for 50S RP and TLR4 were also docked and was utilized as control. The binding affinities and contact interfacesof docked structures at 37 o C were evaluated in PRODIGY web serverwhich uses intermolecular contacts and non-interface surface properties for predictive models (Xue LC, et al., 2016). Binding affinity and binding energy of Covax-TLR4 complex were compared with adjuvant-TLR4 complex. Common residues between the candidate vaccine and the lone adjuvant that are involved in the interaction with human TLR4 were investigated. The crystal structures of HLA-B*53:01 (PDB ID: 1A1M) and HLA-DRB1*04:05 (PDB ID: 4IS6) were retrieved from RCSB database.GalaxyWEBPepDock serverwas utilized to dock the cleaned structures of HLA-B*53:01 and HLA-DRB1*04:05 with two the epitopes having the lowest binding affinity (highest IC50) from each set of selected CTL (FARTRSMWSF) and HTL (TLLALHRSYLT) epitopes resulted in this work. Docked structures were refined in GalaxyWEBRefineComplex server. Binding energy and binding affinity were calculated in PRODIGY web server.
Immune Simulation
The immunogenicity of Covax was further evaluated in silico using the C-ImmSim server. This simulates the cellular and humoral responses of the immune system against an amino acid sequence (Rapin et al., 2010). This tool has been previously used to model Epstein–Barr virus (Castiglione et al. 2007a) and HIV infection (Baldazzi et al. 2006; Castiglione et al. 2007b). Defaultsimulation parameters were used in this study for a prophylactic vaccine injected three times in four weeks intervals (time steps set at 1, 84, and 168) with 200simulation steps. The immune response profile of Covax was compared with that of the adjuvant alone.
In Silico Cloning Optimization of Covax
For the efficient cloning of Covax in E.coli K12 strain, in silico codon optimization was conducted using Java Codon Adaptation Tool (JCAT). This tool calculates for the Codon Adaptation Index (CAI) to approximate the efficiency of gene expression with respect to the subset of highly expressed genes in an organism. The closer the CAI-value to 1, the more a gene will be highly expressed in a host (Grote et al., 2005). GC-content of the candidate vaccine was also calculated.
Results
Identification of Conserved Fragments in S, E, M and N Proteins
Using Shannon entropy threshold < 1.0, the protein variability analysis forretrieved S, E, M, and N sequencesgenerated conserved amino acidsequences(Supp3) for each structural protein which were then usedto predict SARS-CoV-2 epitopes.
Linear BL Epitopes in SARS-CoV-2
Predicted linear BL epitopes from the extravirion sequences of S, E and M proteins yielded1-24 residues. After further assessments, predicted epitopes from E and M proteins did not pass the antigenicity test in Vaxijen server. Only the S protein generated 9 BL epitopes (12-14 residues) whichare all consensus sequences from at least 2 prediction tools. Theseare alsoantigenic, non-allergenic, and without significant matches in human proteins (Table 1).
Table 1: Selected BL epitopes
| Epitope | Antigen | Antigenicity score | Length | Position |
| GTTLDSKTQSLLIV | S | 0.8614 | 14 | 107-120 |
| GAAAYYVGYLQPRT | S | 0.9243 | 14 | 261-274 |
| AVDCALDPLSETKC | S | 0.8534 | 14 | 288-301 |
| GIYQTSNFRVQPTE | S | 1.0038 | 14 | 311-324 |
| GKIADYNYKLPDDF | S | 0.9776 | 14 | 416-429 |
| GFNCYFPLQSYGFQ | S | 0.8237 | 14 | 485-498 |
| YGFQPTNGVGYQ | S | 0.7136 | 12 | 495-506 |
| DQLTPTWRVYSTGS | S | 0.7635 | 14 | 627-640 |
| LSSTASALGKLQDV | S | 0.8753 | 14 | 938-951 |
HTL Epitopes in SARS-CoV-2
Table 2 shows the 12 selected HTL epitopes from S (7), E (1), M (2), and N (2)proteins. All have good binding affinity (IC50 < 500nmol/dm3) withtheir respective human MHC II alleles (Supp4). Furthermore, these 12 epitopes are classified antigenic, non-allergenic, andwithout significant matches with human proteins in the databases.The world population coverage for the series of HTL epitopes in Covax is estimated to be 81.81%, while this set covers 85.83%,
81.82%, 59.99% and 87.89% of populations in Europe, East Asia, Northeast Asia, and North America, respectively. In addition, IPFAMQMAYRF, VTLAILTALRL, SFRLFARTRSM and ALLLLDRLNQL from S, E, M, and N proteins, respectively,may offer cross-protection as they share 100% sequence identity with SARS-CoV besides COVID-19 (Supp5).
Table 2: Selected HTL epitopes
| Epitope | Antigen | Antigenicity
Score |
Position |
| LPFFSNVTWFH | S | 0.7486 | 56-66 |
| PFFSNVTWFHA | S | 0.584 | 57-67 |
| SLLIVNNATNV | S | 0.5504 | 116-126 |
| TLLALHRSYLT | S | 0.5459 | 240-250 |
| LLALHRSYLTP | S | 0.7771 | 241-251 |
| NFRVQPTESIV | S | 1.0669 | 317-327 |
| IPFAMQMAYRF | S | 1.2909 | 896-906 |
| VTLAILTALRL | E | 0.8965 | 29-39 |
| SFRLFARTRSM | M | 0.5944 | 99-109 |
| IGAVILRGHLR | M | 0.8668 | 140-150 |
| PANNAAIVLQL | N | 0.6257 | 151-161 |
| ALLLLDRLNQL | N | 0.5127 | 220-230 |
CTL Epitopes in SARS-CoV-2
Cytotoxic lymphocytes play vital roles against viral pathogens that infect and replicate inside the host cell. This work selected 12 CTL epitopes (9-11 residues) on S (5), E (2), M (4), and N (2) structural proteins of SARS-CoV-2 (Table 3). These epitopes have good binding affinities (IC50 < 500nmol/dm3) withtheir respective human MHC I alleles(Supp6), as well as efficient proteasome processing and TAP scores(>1.0). All 12 epitopes are antigenic, non-allergenic, and without significant matches across human proteins. Besides SARS-CoV-2, these epitopes may offer cross-protection against SARS-CoVas QWNLVIGFLF (M), FARTRSMWSF (M), AQFAPSASAF (N), and IPFAMQMAYRF (S)shared 100% sequence identity with SARS-CoV (Supp5). Moreover, the set of CTL epitopes largely covers 85.87% of world populationwhile 80.17% of population is covered in Northeast Asia particularly China (80.1%), 85.18% in East Asia, 90.78% in Europe, and 86.67% in North America including the U.S.A. with highinfection rates of COVID-19.
Table 3: Selected CTL Epitopes
|
Epitope (residues) |
Antigen | Antigenicity score | Position |
Proteosome -processing score |
TAP score |
| GWTAGAAAYY (10) | S | 0.5358 | 257-266 | 1.24 | 1.31 |
| WTAGAAAYY (9) | S | 0.6306 | 258-266 | 1.24 | 1.24 |
| YYVGYLQPRTF (11) | S | 0.8985 | 265-275 | 1.36 | 1.31 |
| SETKCTLKSF (10) | S | 0.6433 | 297-306 | 1.4 | 1.07 |
| IPFAMQMAYRF (11) | S | 1.2909 | 896-906 | 1.45 | 1 |
| NVSLVKPSFY (10) | E | 0.7279 | 48-57 | 1.19 | 1.36 |
| VSLVKPSFYVY (11) | E | 0.616 | 49-59 | 1.51 | 1.38 |
| QWNLVIGFLF (10) | M | 1.2302 | 19-28 | 1.12 | 1.1 |
| FARTRSMWSF (10) | M | 0.9202 | 103-112 | 1.41 | 1.12 |
| ATSRTLSYY (9) | M | 0.6108 | 171-179 | 1.26 | 1.34 |
| KDLSPRWYFYY (11) | N | 0.9184 | 102-112 | 1.07 | 1.21 |
| AQFAPSASAF (10) | N | 0.5986 | 305-314 | 1.23 | 1.25 |
Construction of Multi-Epitope Vaccine
Figure 1 shows a schematic presentation of Covax multi-epitope construct which consists of 510residues. Herein, the overlapping sequences from the total of 33 selected epitopesgenerated in this work were merged to form21 continuous fragments.Covax includes a Valine residue added at the N-terminus aiming to increase its half-life.
 |
Figure 1: Covax Multi-Epitope Construct |
At the N-terminus of Covax, a Valine residue is bonded to the first residue of 50S RP adjuvant (blue). The adjuvant is linked to the first CTL epitope (maroon) via EAAAK linker (orange). The positions of each epitope based from the sequence of each structural protein is indicated in parenthesis. Five CTL peptide sequences (maroon) were adjoined via AAY linkers (yellow) while 8 HTL (olive green) and 8 BL peptide sequences (pink) were adjoined via GPGPG linkers (violet). BL peptides contain overlapping CTL and HTL epitopes as a result of merging. S, E, M, and N are the four structural antigens of SARS-CoV-2.
Properties of the Vaccine Candidate
Covax is classified as antigenic (0.5852) in Vaxijen server with virus as target, and is also a non-allergen having the highest Tanimoto similarity index of 0.9 with its nearest protein (UniProtKB ID: Q9P281). BlastPanalysis of Covax showed that it has no significant similarity with any human proteins; thus, avoiding possible autoimmune reactions. Protparam tool calculated that Covax has a molecular weight of53,005.45 g/mol, and pI of 7.73 which can be used for purification purposes in isoelectric focusing. Its extinction coefficient is 68,885 M-1 cm-1 at 280 nm wavelength in water, and absorbance is 1.3 assuming all pairs of cysteine residues form cystines. The estimated half-life of Covax is 100 hours in mammalian reticulocytes in vitro, >20 hours in yeast in vivo, and > 10 hours in vivo in E.coli. The instability index is 26.93 which classifies Covax as stable (<40). In addition, an aliphatic index of 81.24 indicates thermal stability while the small positive value of its GRAVY (0.036) implies its very weak hydrophobicity.
Secondary Structure Composition and Disordered Regions inCovax
Figure 2 shows that majority of the secondary structures in Covax are random coils (purple), alpha helix (blue), and extended strands (red). The stretch of linear BL epitopes in Covax(337-510) are mostly random coils and extended strands. In line with these, GlobPlot2 server identified disordered regions in Covax at positions326-342, 352-366, 375-391, 398-415, and 421-504 which are less likely to form stable folded structure making these sequences more flexible for the binding of BCRs. Notice that most of the linear BL peptides in Covax(337-510) are within the disordered regions.
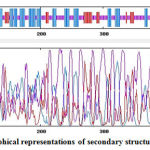 |
Figure 2: Graphical representations of secondary structures in Covax. |
Predicted secondary structures in GOR4 include alpha helices (36.86%) in blue, extended strands (14.90%) in red, and random coils (48.24%) in purple
Prediction, Refinement and Validation of Tertiary Structures
By comparing the quality indicators, initial tertiary structure modelsof Covax, 50S RP and human TLR4 markedlyimproved after refinement (Table 4). Figure 3 shows the best tertiary structure model forCovax.
Table 4: Quality Indicators for Tertiary Structure Validation
| Tertiary structures | ERRAT | Verify3D |
Ramachandran plot (favored, allowed, outlier)
|
| Covax initial | 73.52 % | 76.08 % | 92.6%, 5.2%, 2.2% |
| Covax refined | 80.36 % | 81.57% | 95.5%, 3.5%, 1.0% |
| 50S RP initital | 99.18 % | 84.62 % | 95.3%, 3.1%, 1.6% |
| 50S RP refined | 100 % | 92.31 % | 94.5%, 4.7%, 0.8% |
| TLR4 (PDB:4G8A) | 85.07 % | 96.86 % | 94.4%, 5.1%, 0.5% |
| TLR4 refined | 89.06 % | 97.02 % | 97.7%, 1.8%, 0.5% |
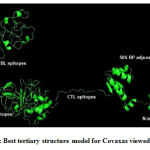 |
Figure 3: Best tertiary structure model for Covaxas viewed inPymol |
Structural B-Cell Epitopes ofCovax
Ellipro revealed 3 linear and 2 discontinuous structural BL epitopesin the tertiary structure of Covax. These structural epitopes overlap with the series of SARS-CoV-2 BL epitopes (337-510) incorporated in the vaccine (Table 5). Structural BL Epitopes in Covax. Linear and discontinuous structural BL epitopes which overlap with the series of SARS-CoV-2 BL epitopes are in bold text.
 |
Table 5: Structural BL Epitopes in Covax. |
Docking of Covax with Human TLR4 and HLA Molecules with Peptides
Figure 4 shows docked structuresof the lowest interaction energyposesfor 50SRP-TLR4 (Fig.4a) and Covax-TLR4 (Fig.4b). The estimated binding energy (ΔG) and binding affinity (Kd) for are -41.42 Kj/mol and 1.0E-07 mol/dm3 for 50SR-TLR4 complex, respectively. While the valuesare -38.49 Kj/mol and 3.4E-07 mol/dm3 for Covax-TLR4. The number of interfacial contacts (IC)per property made at the interface of Covax-TLR4 (IC charged-charged: 20, IC charged-polar: 10, IC charged-apolar: 22, IC polar-polar: 1, IC polar-apolar: 2, IC apolar-apolar: 9) is higher than 50SRP-TLR4 (IC charged-charged: 19, IC charged-polar: 8, IC charged-apolar: 11, IC polar-polar: 5, IC polar-apolar: 11, IC apolar-apolar: 1). To further validate the protocols used in the selection of docked complexes, common interface residues between the two complexes were investigated. GLU16 of Covaxand GLU15 of 50S RP both interact with LYS388 of TLR4. Other common interface residues interacting with TLR4 include GLU9, ALA13, THR18, GLU21 forCovax, and GLU8, ALA12, THR17, GLU20 for 50S RP. Figure 4 also shows that structures of HLA-DRB1*04:05-TLLALHRSYLT (Fig4c) and HLA-B*53:01-FARTRSMWSF (Fig.4d)fitted well on the binding grooves. The binding energy and binding affinity for HLA-DRB1*04:05-TLLALHRSYLT are -49.37 Kj/mol and 5.1E-09 mol/dm3, respectively. While the binding energy and binding affinity for HLA-B*53:01-FARTRSMWSF are -36.40 Kj/mol and 6.9E-07 mol/dm3, respectively.
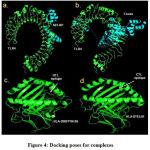 |
Figure 4: Docking poses for complexes. |
ClusProserver generated top poses for 50SRP-TLR4 (a) and Covax-TLR4 (b); and GalaxyWEBPepdock serverHTLepitope-HLADRB1*04:05 (c), and CTLepitope-HLAB*53:01 (d) complexes
Immune Response Profile of Covax
Results from C-ImmSim simulation showed that Covaxcan induce potent immune responses. To prove that the generated immune response is not just because of the presence of 50S RP in Covax but also because the multi-epitopes included in Covax can elicit immunogenic response, immune response from Covax was compared versus the adjuvant alone.The primary response is higher as marked by higher levels of IgMwith Covax than with the adjuvant solely (Figure 5a). The secondary and tertiary responses are greater with Covax as indicated by higher memory BL populations (Fig.5b); and IgG1 + IgG2, IgM, and IgG + IgM levels (Figure 5a).Notice that this trend is also evident with higher HTL (Fig.5c), and CTL (Fig.5d) populations forCovax than when adjuvant is used alone. Cytokine and interleukin production are also indicators of successful immune response from vaccination. Figure 5e shows that Covax induced higher levels of IFN-γ TGF-β, and IL-2 which are important in co-stimulatory signaling for T-cell activation.
 |
Figure 5: C-ImmSim simulation profile with Covax and adjuvant (50S RP) alone induced by 3 injections given 4 weeks apart. |
(a)Immunoglobulin production in response to antigen injections are indicated by black peakswhile specific subclasses areindicated by coloured peaks;(b) development of BL populations per isotype;(c) HTL memory and not memory populations; (d) CTL populations per stateindicate cell at the resting state not presented with the antigen while the anergic state represents tolerance of the T-cells to the antigen due to repeated exposures; and (e) types of cytokines and interleukins induced wherein the insert plot shows IL-2 level with the Simpson index, D (diversity)indicated by the dotted line. Its value is directly proportional to the emergence of diverseepitope-specific dominant clones of T-cellsover time.
In Silico Cloning and Optimization of Covax
The CAI-value for the optimized codons of Covax is 0.95 which is very close to 1. The GC-content, 56.14% is within the optimal range (30%-70%). These results convey that the optimized codons of Covax(1530bp) can be highly expressed in E.coli K12 strain as a host.
Discussion
SARS-CoV-2hashigher transmission rate compared with other deadlier coronaviruses, SARS-CoV, and MERS-CoV(Petrosillo et al., 2020). Though it has lower case fatality rate (2.3%),its casualties can be further magnified by its highly contagious nature. Development of vaccines, anti-viral drugs and studies on biological properties of SARS-CoV-2 have been the main focus of researchers around the world since the emergence of COVID-19 pandemic. The most common practices for the prevention of disease-causing viral infections include the use of live attenuated, whole inactivated, or purified antigen. Live attenuated vaccines are often immunogenic; however, some live attenuated vaccines have high risk of virulent virus reversion especially in immunosuppressed individuals. Inactivated vaccines consist of whole or purified antigens, contain components that may cause unwanted adverse effects and tolerability concerns (O’Hagan et al., 2001).The rapid increase in the number of SARS-CoV-2-infected individuals worldwide warrants the development of effective measures to counteract its infection in an accelerated manner.Epitope-based vaccines has gainedattention as it offers more potential advantages over whole purified antigen. This approach focuses the immune responses on specific antigenic determinants recognized by immune receptors of lymphocytes while minimizing the undesirable immune reactions from the whole antigen. With the advancements in immunoinformatics, in silico approach is currently employed to successfully discover multi-epitope vaccines against various pathogens (Oli et al., 2020).
Because oftheir expressionabundance, important roles in viral pathogenesis and replication,all the structural proteins of SARS-CoV-2 were utilized for the screening of epitopes in this work. The protocol used in this study generated 9 linear BL epitopes which are all from S protein. Two antigenic epitopes GFNCYFPLQSYGFQ (485-498) and YGFQPTNGVGYQ (495-506) overlap with the receptor binding motif (RBM) (437-508) site of S protein. RBM is a region in the S protein which binds to angiotensin converting enzyme 2 (ACE2) receptor of the host cell (Letko et al., 2020). Upon binding of antibodies to these epitopes on RBM, the attachment of SARS-CoV-2to the host cell can be hindered. Studies showed that antibodies are short-lived in convalescent patients compared to HTL and CTL responses which can provide long-term immunity even after a decade of SARS-CoV infection (Channappanavar et al., 2014). Thus, besideslinear BL epitopes, T-cell epitopes were also included in the vaccine construct. Activation of BL using epitope-based vaccines requires presentation of MHCII-peptide complex to an activated HTL. One of the major challenges in the development of peptide vaccines is the MHC haplotype-restricted antigen recognition. Thus, this work generated 12 HTL and 12 CTL epitopes with good binding affinity towards the most frequent MHC II and MHC I alleles to cover a large population. A total of 33 epitopes which consist of 9 BL (12-14 residues), 12 HTL (11 residues), and 12 CTL (9-11 residues) epitopes were generated using this protocol. Generally,MHC I moleculesbind to peptides with 9-11 residues while MHC II-bound peptides vary from 9–22 residues (Sanchez-Trincado et al. 2017).The approach of this work increasesthe chance of peptide presentation as all four structural antigens havecorresponding HTL and CTL epitopes.Even prior to the incorporation of epitopes in the multi-epitope construct, in silico analysisclassified all epitopes included in the vaccine as non-allergen whichreduces potential allergic reactions. The E-values of epitopes blasted against human protein databases are greater than 1. Matches with E-values smaller than 1.0E-30 can be cross-reactive in some allergic individuals (Hileman et al., 2002). Thus, these epitopes are less likely to cause autoimmune reactions in humans.Given that all selected epitopes are included in the vaccine, results showed large world population coverage estimated for the set of HTL (81.81%) and CTL (85.87%) epitopes as well as in areas where infection rates are high. Sincesome MHC alleles for the sets of HTL and CTL epitopes were not covered byIEDB-PC tool, the estimated population coverage is minimum—the actual percentagecan be larger. Having > 80% population coverage, this candidate vaccine may provide herd immunity against SARS-CoV-2 in many populations around the world. In addition, cross-reactivity analysis with 7 HCoVs showed that several HTL epitopes IPFAMQMAYRF (S), VTLAILTALRL (E), SFRLFARTRSM (M), and ALLLLDRLNQL (N); and CTL epitopes QWNLVIGFLF (M), FARTRSMWSF (M), AQFAPSASAF (N), and IPFAMQMAYRF (S) shared 100% sequence identity with SARS-CoV which may offer cross-protection against this virus besides COVID-19.
The form and stability of vaccines are important factors to be considered to ensure efficient delivery of targeted actions. This work focused on developing a multi-epitope vaccine over the use of single epitopes. Advantages include simultaneous induction of immune response as a result of adjoined promiscuous epitopes, linked adjuvant, and exclusion of sequences that may cause unwanted effects. Epitopes were adjoined using AAY and GPGPG linkers which were proven to present sufficient epitopes in vivo (Jin et al., 2009; Negahdaripour et al., 2018). While EAAAK linker was used to preserve the bioactivity of adjuvant in Covax(Arai et al., 2001).Incorporating adjuvants in vaccines is essential to be able to enhance its immunogenicity (Lei et al., 2019). This workadjoined the multi-epitopes with 50S RP from M.Tuberculosisas an adjuvant since it has been known to bind and activate TLR4. Studies showed that 50S RP induces increased expression of dendritic cell maturation markers, T-cell activation, and cytokines in a TLR4-dependent manner (Lee et al., 2014; Kim et al., 2012). The evaluation of antigenicity and allergenicityof Covaxconstructclassified it as antigenic and non-allergenic, emphasizing its potential and safety as an immunotherapeutic agent.To add to its safety profile, BlastPanalysis of showed that Covax has no significant matches across human protein databases,avoidingpossible autoimmune reactions.In silico evaluations of the physicochemical properties of Covax proved its stability. Protein degradation is often estimated in its amino-terminal residue; thus, Valine was added at the N-terminus of the constructimproving its half-life from 30 hours (without Val) to 100 hours (with Val) in mammalian reticulocytes. In addition, the instability index of this candidate vaccine is <40 which implies longer protein half-life in vivo based on its dipeptide composition (Guruprasad et al., 1990). The antigenicity of BL epitopes in a vaccine may also depend onwhether they are exposed in the structure of the vaccine so BCRs can easily interact. Results showed that the secondary structures of the series oflinear BL epitopes in Covax (337-510) are random coils and extended strands. These epitopes are also found within the disordered regions of the vaccine. Disordered regions in proteinshave less tendency to form stable folded structures and are more flexible; therefore, these BL epitopes are more exposed for the binding of BCRs. Several indicators validated the quality of tertiary structuresused in this work. Based from the study of Singh and colleagues (2016), any modelled structure exhibiting ERRAT score value > 50 is considered good. In Ramachandran plot analysis, a model with good quality is expected to have at least 90% of residues within the most favored region (Laskowski et al., 1993). Good structures have at least 80% of residues with 3D-1Dscore≥ 0.2 (Eisenberg et al., 1997). Evaluations using these quality indicators showed that the improved quality of refined structures versus initial structures justified the need for refinement. These quality indicators also validated the tertiary structure models used for Covax, 50S RP and TLR4 in this study. The antigenicity of this candidate vaccine is further supported by the resulting linear and discontinuous structural BL epitopes in Covax based from the results inEllipro. And to prove not just the antigenicity but also immunogenicity, data-driven docking was conducted to determine possible immune interaction between the vaccine and TLR4. This immune receptor was used in the analysis because Covax contains 50S RP which is a known TLR4-agonist. Knowing that 50S RP binds to TLR4 in vivo, the values of the binding parameters for 50SRP-TLR4 complex can be used a control. Results from this work showed that the binding energy and binding affinity of Covax-TLR4, is very close to that of 50SRP-TLR4. These values also indicate that the binding of Covax to TLR4 is spontaneous (negative ΔG) and that the formation of Covax-TLR4 complex is favoured (Kd<<1), similar to that of TLR4 with 50S RP alone.Because 50S RP is known to interact with TLR4 in vivo; therefore, Covax is expected to interact with TLR4 on immune cells, such as dendritic cells, resulting to development of protective immune response against SARS-CoV-2.
The methods and tools used to select CTL and HTL epitopes were further validated by docking peptides having the lowest binding affinity (highest IC50) with their corresponding MHC alleles. Calculated binding affinities from Prodigy server (Kd)indicate that the binding of each epitope with its MHC allele is favored (Kd< 1) andthat the formation of HLA-DRB1*04:05-TLLALHRSYLT and HLA-B*53:01-FARTRSMWSF complexes are spontaneous and stable as indicated by negative ΔG values. This step provided more compelling evidence on the accuracy of the T cell prediction methods used in this work—if the tested peptides with the lowest binding affinities (highest IC50) can bind favourably and stably with their corresponding MHC alleles, more so those epitopes in the list with higher binding affinities (lower IC50).
Immune simulationplots forCovax showed that it can induce immune responses. Notice that the levels of IgG, IgM antibodies, memory BL, memory HTL and activated CTL are induced, supporting evident humoral responses and long-term memory persistence after three times exposure with the candidate vaccine. The induced immunogenicity is also evident in the efficacy of antigen clearance from the subsequent injections of the vaccine. Cytokine simulation plot showed increased IFNγ and IL-2 with Covax similar to the clinical features observed in SARS-CoV-2-infected patients (Huang et al., 2020). More importantly,the immune responses withCovaxis higher thanwith 50S RP adjuvant alone (Fig.5).For example, the level of primary response IgM antibody titer for Covax (100,000) is much greater than with the adjuvant alone (60,000) (Fig.5a). Notice that IFN-γ is highly maintained throughout exposure with Covax but was reduced over time with the exposure to adjuvant alone (Fig.5e). With this comparison, it is evident that the generated immune responses are not just because of the presence of 50S RP in Covax but also because the multi-epitopes incorporated in Covax(BL, HTL & CTL) can elicit immunogenic responses. The GC content and high codon adaptability index of the optimized codons for Covax recombinant vaccine suggest favorable high level expression in E. coli (strain K12).
Results of this work on the potential immunogenicity and safety of Covax were all generated in silico. Further studies on the immunogenicity, efficacy, and possible adverse effects should be conducted both in vitro, and in vivo. Future studiescould include the expression of this candidate vaccine in bacterial system to screen for immunoreactivity through serological analysis.
Conclusion
Immunoinformatics approach can aid in designing safe and effective prophylactic agents with lesser time requiredforsituations such as COVID-19 pandemic. This is the first work to design a vaccine containing multi-epitopes against all the structural proteins ofSARS-CoV-2. Results showed that Covax confers stability, safety and contains epitopes that are antigenic, and immunogenic. Nonetheless, the application of Covaxas a candidate vaccine is anticipated to be authenticated both in vitro and in vivo.
Acknowledgement
I would like to thank the reviewers who shared helpful insights to this manuscript.
Conflict of Interest
I have no conflicts of interest to be declare.
Funding Source
There are no funding source .This work is self-funded.
References
- Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses, 2020; 12(3): 254.
CrossRef - Al-Tawfiq JA, Zumla A, Memish ZA. Travel implications of emerging coronaviruses: SARS and MERS-CoV. Travel Medicine and Infectious Disease, 2014; 12(5): 422–428.
CrossRef - Arai R, Ueda H, Kitayama A, et al. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng., 2001; 14: 529-32.
CrossRef - Baldazzi V, Castiglione F, Bernaschi M. An enhanced agent based model of the immune system response. Cell Immunol., 2006; 244, 77-79.
CrossRef - Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics, 2006; 7: 153.
CrossRef - Castiglione F, Duca K, Jarrah A, et al. Simulating Epstein–Barr virus infection with C-ImmSim. Bioinformatics,2007a; 23, 1371-1377.
CrossRef - Castiglione F, Pappalardo F, Bernaschi M, Motta S. Optimization of HAART with genetic algorithms and agent-based models of HIV infection. Bioinformatics,2007b;23, 3350-3355.
CrossRef - Channappanavar R, Fett C, Zhao J, Meyerholz DK, Perlman S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J Virol., 2014; 88(19): 11034-44.
CrossRef - Colovos C and Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci., 1993; 2: 1511-9.
CrossRef - Dimitrov I, Naneva L, Doytchinova I, et al. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics, 2014; 30: 846-51.
CrossRef - Doytchinova IA and Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics., 2007; 8:
CrossRef - Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol., 1997; 277: 396‐
CrossRef - Emini EA, Hughes JV, Perlow DS, et al. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide.J Virol., 1985; 55: 836-9.
CrossRef - Galbadage T, Peterson B, Gunasekera R. Does COVID-19 Spread Through Droplets Alone? Frontiers in Public Health, 2020; 8: 163.
CrossRef - Greenbaum J, Sidney J, Chung J, et al. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics, 2011; 63: 325-35.
CrossRef - Grote A, Hiller K, Scheer M, et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res., 2005; 33: W526-31.
CrossRef - Guruprasad K, Reddy BV, Pandit MW, et al. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng., 1990; 4: 155-61.
CrossRef - Hileman RE, Silvanovich A, Goodman RE, et al. Bioinformatic methods for allergenicity assessment using a comprehensive allergen database. Arch. Allergy Immunol., 2002; 128: 280–291.
CrossRef - Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell, 2020; 181(2): 271-280.
CrossRef - Hoof I, Peters B, Sidney J, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics, 2009; 61(1): 1‐
CrossRef - Hu Y, Wen J, Tang L, et al. The M protein of SARS-CoV: basic structural and immunological properties. Genomics Proteomics Bioinformatics, 2003;1(2):118‐ doi:10.1016/s1672-0229(03)01016-7.
CrossRef - Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet, 2020; 395(10223): 497‐
CrossRef - Jensen KK, Andreatta M, Marcatili P, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology, 2018; 154(3):394‐
CrossRef - Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Research, 2017; 45(W1): W24–9.
CrossRef - Jin X, Newman MJ, De-Rosa S, et al. A novel HIV T helper epitope-based vaccine elicits cytokine-secreting HIV-specific CD4+ T cells in a Phase I clinical trial in HIV-uninfected adults.Vaccine, 2009; 27: 7080-6.
CrossRef - Kim K, Sohn H, Kim J-S, et al. Mycobacterium tuberculosis Rv0652 stimulates production of tumour necrosis factor and monocytes chemoattractant protein-1 in macrophages through the Toll-like receptor 4 pathway. Immunology, 2012; 136(2): 231–240.
CrossRef - Ko J,Park H, Seok C. GalaxyTBM: template-based modeling by building a reliable core and refining unreliable local regions. BMC Bioinformatics, 2012; 13: 198.
CrossRef - Kolaskar AS and Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett., 1990; 276: 172-4.
CrossRef - Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking. Nature Protocols, 2017; 12(2): 255-278.
CrossRef - Kuo L, Koetzner CA, Masters PS. A key role for the carboxy-terminal tail of the murine coronavirus nucleocapsid protein in coordination of genome packaging. Virol., 2016; 494: 100–107.
CrossRef - Laskowski RA, MacArthur MW, Moss DS,et al. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 1993; 26: 283-91.
CrossRef - Lee SJ, Shin SJ, Lee MH, et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS ONE, 2014; 9: e104351.
CrossRef - Lei Y, Zhao F, Shao J, et al. Application of built-in adjuvants for epitope-based vaccines. PeerJ, 2019; 6: e6185.
CrossRef - Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol, 2020; 5(4): 562-569.
CrossRef - Li CK, Wu H, Yan H, et al. T cell responses to whole SARS coronavirus in humans. J Immunol, 2008; 181(8): 5490-500.
CrossRef - Li Q, Guan X, Wu P, et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N Engl J Med, 2020; 382: 1199-1207.
CrossRef - Ma Y, Tong X, Xu X, Li X, Lou Z, Rao Z. Structures of the N- and C-terminal domains of MHV-A59 nucleocapsid protein corroborate a conserved RNA-protein binding mechanism in coronavirus. Protein Cell, 2010; 1(7): 688-697.
CrossRef - McBride R, van Zyl M, Fielding BC. The coronavirus nucleocapsid is a multifunctional protein. Viruses, 2014; 6(8): 2991‐
CrossRef - Negahdaripour M, Nezafat N, Eslami M, et al. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infect. Genet. Evol, 2018; 58: 96–109.
CrossRef - Neuman BW, Kiss G, Kunding AH, et al. A structural analysis of M protein in coronavirus assembly and morphology. J StructBiol, 2010; 174: 11–22.
CrossRef - Nieto-Torres JL, De Diego ML, Verdiá-Báguena C, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PloSPathog, 2014; 10(5): e1004077.
CrossRef - O’Hagan DT, MacKichan ML, Singh M. Recent developments in adjuvants for vaccines against infectious diseases. BiomolEng, 2001; 18: 69-85.
CrossRef - Oli AN, Obialor WO, Ifeanyichukwu MO, et al. Immunoinformatics and Vaccine Development: An Overview. Immunotargets Ther, 2020; 9: 13-30.
CrossRef - Oyarzún P, Kobe B. Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Human Vaccines &Immunotherapeutics, 2016; 12(3):763-767.
CrossRef - Pan M, Wang X, Liao J, et al. Prediction and identification of potential immunodominant epitopes in glycoproteins B, C, E, G, and I of herpes simplex virus type 2. Clinical & Developmental Immunology, 2012; 2012: 205313.
CrossRef - Petrosillo N, Viceconte G, Ergonul O, Ippolito G, Petersen E. COVID-19, SARS and MERS: are they closely related?.ClinMicrobiol Infect, 2020; 26(6): 729‐
CrossRef - Pleguezuelos O, James E, Fernandez A, et al. Efficacy of FLU-v, a broad-spectrum influenza vaccine, in a randomized phase IIb human influenza challenge study. NPJ Vaccines, 2020; 5: 22.
CrossRef - Ponomarenko J, Bui H,Li W, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics, 2008; 9: 514.
CrossRef - Rapin N, Lund O, Bernaschi M, Castiglione F, et al. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE, 2010; 5(4): e9862.
CrossRef - Saha Sand Raghava GP. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins, 2006; 65, 40-8.
CrossRef - Sanchez-Trincado JL, Gomez-Perosanz M, Reche PA. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J Immunol Res, 2017; 2017:2680160.
CrossRef - Shannon C. The mathematical theory of communication. Bell Syst Tech J, 1948; 27: 379–423.
CrossRef - Singh S., Singh D.B., Singh A., Ram B.G.G., Dwivedi S., Ramteke P.W. An approach for identification of novel drug targets in Streptococcus pyogenes SF370 through pathway analysis. Sci. Comput. Life Sci,2016; 8: 388–394.
CrossRef - Vaishnav N, Gupta A, Paul S, John GJ. Overview of computational vaccinology: vaccine development through information technology. J Appl Genet, 2015; 56(3):381‐
CrossRef - Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, Dediego ML, Enjuanes L, Aguilella VM. Analysis of SARS-CoV E protein ion channel activity by tuning the protein and lipid charge. BiochimBiophysActa, 2013; 1828: 2026–2031.
CrossRef - Walls AC, Tortorici MA, Bosch BJ, et al. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature, 2016; 531(7592):114‐
CrossRef - Wang D, Hu B, Hu C, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA, 2020; 323(11): 1061–1069.
CrossRef - Weiskopf D, Angelo MA, de Azeredo EL, et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc Natl AcadSci U S A, 2013; 110(22): E2046-E2053.
CrossRef - Wilson L, McKinlay C, Gage P, Ewart G. SARS coronavirus E protein forms cation-selective ion channels. Virology, 2004; 330:322–331.
CrossRef - Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics, 2016; 32(23) :3676‐
CrossRef - Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 2020; 579(7798): 270‐
- Zhu X, Liu Q, Du L, Lu L, Jiang S. Receptor-binding domain as a target for developing SARS vaccines. J Thorac Dis., 2013; 5 Suppl 2(Suppl 2): S142-S148.

