
Manuscript accepted on :05-Sep-2020
Published online on: --
Plagiarism Check: Yes
Reviewed by: Ismail Abdullahi
Pallavi M S and Pramod Kumar H S
Department of Computer Science, Amrita School of Arts and Sciences, Amrita Vishwa Vidyapeetham, Mysuru Campus, India.
Corresponding Author E-mail: palls.ms@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/2018
Abstract
To assess an effective medication against BRAF(V600E), the gene which causes melanoma skin cancer , a literature survey was performed and three drugs were identified that are effective melanoma cell inhibitors that induce skin cancer and help patients cure skin cancer. In this analysis we analyzed these three drugs namely Aknadicin as (Drug1) and 16beta-hydroxy-vendolinine-n-oxide as (Drug2) and PID (215) pronounced (1Z)-5-(2-{ 4 - [dimethylamino) ethoxy] phenyl }-5- pyridin -4-yl-1h- imidazol- ylindian-1- oneoxime as (Drug3) using various methodologies and simulation techniques to find out how highly successful these drugs are among them. In this research, we developed the 3D structure of BRAF(V600E) through homology modeling and validated it through Ramachandran plot and verify3D and implemented the method of docking through virtual screening of protein with drug and simulation of molecular dynamics to further suggest the powerful potent novel inhibitors for malignant melanoma management. Obtained a wild form BRAF(V600E) 3D structure and docked protein structure with each drug and continued molecular dynamic simulation by measuring the stability and energy interaction between the protein and these drugs. An RMSD graph displays the drug’s efficacy. Analysis of three different drugs using computational techniques by performing different methodologies of docking, molecular simulation and interaction energy and results of each drug in these methodology, reveals the efficiency of drugs. Based on the results we identify that the Drug3 is a highly effective inhibitor of melanoma among other drugs.
Keywords
BRAF inhibitor; Charge calculation; Docking; Homology modelling; Interaction energy; Molecular dynamic simulation; Structure-based; Virtual screening
Download this article as:| Copy the following to cite this article: Pallavi M. S, Kumar H. S. P. In-silico Analysis to Determine the Efficient Drug for Malignant Melanoma using Molecular Dynamics. Biomed Pharmacol J 2020;13(3). |
| Copy the following to cite this URL: Pallavi M. S, Kumar H. S. P. In-silico Analysis to Determine the Efficient Drug for Malignant Melanoma using Molecular Dynamics. Biomed Pharmacol J 2020;13(3). Available from: https://bit.ly/3jvawgh |
Introduction
Melanoma is the skin-cancer that arises in the cells through gene mutation and grows uncontrolled or advanced metastatic cells. BRAF(V600E) is the gene that causes skin cancer of melanoma when amino acid valine(V) is substituted by glutamic acid(E) at the 600th position of the amino acid sequence, whereas the protein found in BRAF transforms it into wild BRAF that causes skin cancer.1 This transition has led to the constitutive activation of RAS mutation on the MAPK pathway.2 In patients with malignant melanoma who have a single mutation, use of BRAF inhibitors has become an exciting therapeutic option. One of the first options to treat metastatic melanoma was that of BRAF inhibitors.1 Treatment choices for patients with metastatic melanoma have been through in recent years but the overall survival rate has so far failed to increase. So, this affects innovative and successful methods to discover and design.3
Various medications have been proposed for melanoma such as ipilimumab and vemurafinab,2 aknadicin, 16-beta- hydroxy- vendolinine-n-oxide1 and 215 (Drug3),3 yet none of these have been evaluated. Based on the molecular behavioral research, the computational methods have been proposed for in-silico study of cancer dynamics to help us compute model and compare these drugs.4 In this study, we analysed and compared the three potential inhibitors of melanoma cells where two novel inhibitors Drug1 and Drug2 are considered1 as they were found efficient drugs against melanoma cells and another drug inhibitor present in the protein complex with protein id 2FB8 namely 215(Drug3)3 are compared with the computational methodologies as explained in the later stages. The protocols used are as same as approaches used by Chen et.al.2015.
In this pipeline, we mainly follow three methodologies: firstly homology modelling of the protein structure to obtain 3D- structure of the protein, further evaluating the 3D-structure with the construction of Ramachandran plot with rampage http://mordred.bioc.cam.ac.uk/~rapper/rampage.php and verify3D.5 Secondly, docking was performed for the predicted protein structure along with the three drug components [mentioned above] to check their interaction with the protein by analysing binding affinity. Thirdly, molecular dynamics (MD) simulation was performed for analysing the physical movements of atoms and molecules. MD method is frequently applied to study the motions of macromolecules such as proteins and nucleic acids, which can be useful for analysing modelling interactions with other molecules as in ligand docking. Total interaction energy, root mean square deviation, root mean square fluctuation were also calculated in this method.
Materials and Methods
Retrieval of Drugs
We have considered three drugs in this study for comparison, to find the optimal drug for wild type BRAF. The drug aknadicine(drug1) pubchem ID (442156)4 was downloaded in the form of SDF format, later converted into PDB format from Obabel 6 The drug 16beta-hydroxy-19s-vindoline N-oxide(drug2) with external ID (5318348) as 3D structure was not available, first we have obtained 2D structure with its ID by drawing in mobile molecular model application from google play store and visualised in discovery studio7 to obtain 3D structure we retrieve the structure in PDB format. The third Drug3 was retrieved from RCSB8 where protein name 2FB8 with the PID 215.3
Homology modelling
A human BRAF obtained the UniProt Knowledgebase (P15056, human), the sequence of it with its residues 1–766 and 3D structure residues 445–723 from RCSB Protein ID 2FB8.8 The homology modeling system for BRAF(V600E) with wild-type crystal structure BRAF (2FB8) on Modeller Software9,10,8,11 was created. When modeling homology we first replace the amino acid valine(V) with glutamic acid (E) at position 600 in the BRAF protein sequence and create the BRAF(V600E)-modelled structure based on the wild-type BRAF (2FB8, human) prototype. Also the structure was constructed through Phyre software9 and was compared through chimera UCSF.10 Further, the obtained protein structure from modeller software was validated using Ramachandran plot using Rampage and verify3D.5
Molecular Docking and Charge Calculation
Molecular Docking was performed using mgl tools11 and AutoDock Vina12 using Lamarckian genetic algorithm [LGA] with 25,000,000 steps. The grid parameters were set to size 60, 64, 54 with centers 16.904, -0.229 and 56.704 for all three drugs. The resultant docked structure was visualized through Chimera and polar-coordinates of the docked images were visualised through Pymol.13 The charge for the drug was calculated using Resp Esp charge Derive Server(REDS) 14 which is used to generate force field parameters using AMBER.
Molecular Dynamics and Binding Energy Estimation
Molecular dynamics (MD) simulation was performed using Gromacs 5.1.4 15,16 Amber99 force field was applied to generate the gromacs topology for protein, AMBER1617 for used to check the parameters of the drug and acpype18 was used to generate gromacs topology for drug. A dodecahedron box was used with extended simple point charge water molecule19 Energy minimization was performed for 50,000 steps using the steepest descent minimization process. Simulation of the system’s position-restrained dynamics (equilibration phase) (NVT and NPT) at 300 K for 200 ps followed by MD production run for 10 ns. The plot for the RMSD was produced with Xmgrace.20 The 3D structures were generated using Chimera and visualized using VMD.2
Results
Constructing 3D structure of protein using homology modelling
The BRAF(V600E) protein 3D structure was modeled using Modular Software as shown in Figure 1(c). The structure was validated using the Ramachandran plot Figure 1(a), the model structure showed that 82.7% of residues were in the preferred region, 13.1% were in the permissible area, and just 4.1% were in the outer area. Verify3D score indicated that the BRAF(V600E)-modelled structure is an ideal conformation model. Verify3D score of BRAF(V600E) protein showed that almost all the residues had positive values Figure 1(b).
 |
Figure 1: (a) Ramachandran plot of BRAF protein structure. (b) Verify3D score of BRAF protein. (c) BRAF(v600e) 3D protein structure. |
Docking or Virtual Screening
A structural docking of all the drugs was performed with BRAF(V600E) protein. The Drug1, Drug2 and Drug3 binding affinity score was -8.1, -8.4 and -8.8 kcal/mol respectively as shown in Table 1. The docked 3D structure of the drugs with protein is shown in Figure 2 (b, d and f) of the three drugs represented in Figure 2 (a, c and e) docked with the BRAF(V600E), respectively. Binding affinity scores shows that Drug3 has a higher binding affinity with the protein. We further proceeded to MD simulation to find the stability of these drugs with the protein.
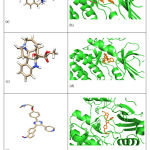 |
Figure 2: (a) 3D structure of aknadicin drug as (Drug1). (b) BRAF protein docked structure of protein with Drug1. (c ) 3D structure of 16beta-hydroxy-vendolinine-n-oxide drug as (Drug2) (d) BRAF protein docked structure of protein with Drug2. (e) 3D structure of (215) (Drug3) (f) BRAF protein docked structure of protein with Drug3. |
Table 1: Binding affinity score of all three drugs with BRAF protein.
| Drug Name | Docking Score |
| Aknadicin | -8.1 kcal/mol |
| 16beta-hydroxy-vendolinine-n-oxide | -8.4 kcal/mol |
| (215) Drug3 | -8.8 kcal/mol |
Molecular Dynamic(MD) Simulation
The 10ns MD simulation of BRAF protein with each drug complex is carried out using AMBER16 forcefield, the gromacs topology is applied from protein , the REDS(red server) is used to calculate drug topology and is validated using antechamber. The energy minimization protocol is used in the SPC water model. The stability of the BRAF- drug complex is computed using RMSD as shown in Figure 3(a). From the result, it is revealed that the BRAF- Drug3 complex was more stable than the BRAF- Drug1 and BRAF-Drug2 complex.
To know the fluctuation of the protein with its drug, RMSF was computed as shown in Figure 3(c). Drug3 indicates less fluctuation. To know the measure of compactness of the protein, the radius of gyration is calculated as represented in Figure 3(b). With Figure 3(b), we can know that protein with Drug3 is more compact than other two complexes.
Further, the interaction energy of all the three complexes, BRAF-Drug1 is -210.0979 +/- 4.8 kJ/mol, BRAF-Drug2 is -176 .838 +/- 2.5 kJ/mol and BRAF-Drug3 is -226.9318 +/- 8.2 kJ/mol which is as represented in Table 2. The interaction energy also confirms that BRAF-Drug3 is having low interaction energy than other two complexes.
 |
Figure 3: (a) RMSD of BRAF protein with its three drugs (b) Radius of Gyration of BRAF protein with its three drugs (c) RMSF of BRAF protein with its three drugs. |
Table 2: Interaction energy of all three drugs with its BRAF protein
| Drug Name | Interaction energy |
| Aknadicin | -210.0979 +/- 4.8 kJ/mol |
| 16beta-hydroxy-vendolinine-n-oxide | -176 .838 +/- 2.5 kJ/mol |
| (215) Drug3 | -226.9318 +/- 8.2 kJ/mol |
Discussion
Through literature survey, we have identified top three drugs namely aknadicin and 16beta-hydroxy-vendolinine-n-oxide referred as Drug1 and Drug2 respectively and third drug 215 (Drug3) was retrieved from RCSB where Protein ID 2FB8 with the Drug ID 215.
In this study, we are identifying the most efficient drug or inhibitor to melanoma cells causing skin cancer by comparing these three drugs using computational analysis. Here, we constructed the 3D structure of a wild-type BRAF(V600E) protein using homology modelling and validated it through the Ramachandran plot and verify3D. Then we perform structural docking of protein with each drug and calculate the binding affinity score. Binding affinity score of Drug1, Drug2 and Drug3 was -8.1 kcal/mol, -8.4 kcal/mol, and -8.8 kcal/mol respectively. From the result, it is revealed that the BRAF-Drug3 complex has higher binding affinity than the BRAF-Drug1 and BRAF-Drug2 complex.
We further proceeded to MD simulation to analyze stability of these drugs with protein and to know the conformational changes of the drug with protein. Results of RMSD,RMSF and radius of gyration indicate that the BRAF-Drug3 complex is more stable than the BRAF-Drug and BRAF-Drug2 complex. The computed interaction energy of all the three complexes, BRAF-Drug1 is -210.0979 +/- 4.8 kJ/mol, BRAF-Drug2 is -176 .838 +/- 2.5 kJ/mol and BRAF-Drug3 is -226.9318 +/- 8.2 kJ/mol also revealed that Drug3 is more efficient that other two drugs. Thus from the result of docking, MD simulation, interaction energy calculation it is concluded that Drug3(215) is the most efficient drug or potent inhibitor of melanoma cells among three drugs.
Conclusion
On the basis of results of homology modelling, structure-based virtual screening or docking, MD simulation and interaction energy calculation of protein and drug complex , we recommend Drug3 (215 pronounced as (1Z) -5- (2-{4-[dimethylamino) ethoxy]phenyl}-5- pyridin -4-yl-1h- imidazol-ylindian-1- one-oxime) as a more efficient drug or potent inhibitor of melanoma cells causing skin cancer.
Financial support and sponsorship
Nil
Conflicts of interest
There are no conflicts of interest.
References
- Tang H-C, Chen Y-C. Insight into molecular dynamics simulation of BRAF(V600E) and potent novel inhibitors for malignant melanoma. Int J Nanomedicine 2015;10:3131–46.
- Zhou L, Yang K, Andl T, Wickett RR, Zhang Y. Perspective of Targeting Cancer-Associated Fibroblasts in Melanoma. J Cancer 2015;6:717–26.
- King AJ, Patrick DR, Batorsky RS, Ho ML, Do HT, Zhang SY, et al. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res 2006;66:11100–5.
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 2019;47:D1102–9.
- Eisenberg D, Lüthy R, Bowie JU. [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods in Enzymology, vol. 277, Academic Press; 1997, p. 396–404.
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform 2011;3:33.
- Studio BD. Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment [Internet] 2016.[cited 2016 August 14] Release 2017, San Diego: Dassault Systèmes, 2016 n.d.
- Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr 2002;58:899–907.
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015;10:845–58.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12.
- Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 2016;11:905–19.
- Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ. Virtual Screening with AutoDock: Theory and Practice. Expert Opin Drug Discov 2010;5:597–607.
- DeLano WL, Others. Pymol: An open-source molecular graphics tool. CCP4 Newsletter on Protein Crystallography 2002;40:82–92.
- Vanquelef E, Simon S, Marquant G. RED Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids 2011.
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem 2005;26:1701–18.
- Lemkul J. From proteins to perturbed Hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package [article v1. 0]. Living Journal of Computational Molecular Science 2018.
- Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. Journal of 2004.
- Sousa da Silva AW, Vranken WF. ACPYPE – AnteChamber PYthon Parser interfacE. BMC Res Notes 2012;5:367.
- Mark P, Nilsson L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J Phys Chem A 2001;105:9954–60.
- Turner PJ. XMGRACE, Version 5.1. 19. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR 2005.
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996;14:33–8, 27–8.

