
Sumitha A1*, Parthiban Brindha Devi2, Sowmya Hari 3 and Dhanasekaran R4
1Department of Pharmacology, Acs Medical College, Chennai,India -600077
2Head of The Department, Department of Biotechnology, School of Engineering, Vels University, Chennai- 600043
3Department of Paediatrics, Chennai, India -600003
Corresponding Author E-mail : arum.sumithadr@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/1986
Abstract
Coronavirus disease (covid-19) is a pandemic of international concern. It creates serious health risk all around the globe and it has no effective treatment. Doxycycline and Quinine are the drugs used as ligands in the study as these drugs has proved in vitro antiviral activity against dengue virus and herpes simplex virus. These compounds were targeted against non structural protein (nsp 12) which plays a vital role in replication and transcription of Corona viral genome. The protein 6 NUR that showed maximal identity of target protein nsp 12 was retrieved using BLASTp. Further the protein was modelled and the compounds were docked using AUTODOCK software and to study the structure activity relationship of ligand with target protein and biochemical information of ligand receptor interaction was done. Both the compounds, Doxycycline and Quinine were well engaged into the active site of target protein nsp 12 with strong hydrogen bond interaction and non polar interaction with active site of the protein. The Docking score of Doxycycline is found to be - 7.34 kcal/mol while that of Quinine being -6.14 Kcal/mol. This indicates the potential of these drugs as a lead against the nsp target protein of Corona virus which need further analysis and Optimisation studies.
Keywords
Corona virus; Docking; Doxycycine; In silico; Quinine
Download this article as:| Copy the following to cite this article: Sumitha A, Devi P. B, Hari S, Dhanasekaran R. Covid-19 - In Silico Structure Prediction and Molecular Docking Studies with Doxycycline and Quinine. Biomed Pharmacol J 2020;13(3). |
| Copy the following to cite this URL: Sumitha A, Devi P. B, Hari S, Dhanasekaran R. Covid-19 - In Silico Structure Prediction and Molecular Docking Studies with Doxycycline and Quinine. Biomed Pharmacol J 2020;13(3). Available from: https://bit.ly/3mcF881 |
Introduction
Corona virus belongs to positive stranded RNA virus which is enveloped. Severe acute respiratory syndrome coronavirus 2 (SARS CoV -2 or COVID 2019) was reported first in Wuhan,China in December 2019.1 Later it has spread rapidly to other countries around the world. Fever, cough, shortness of breath and dyspnea are common symptoms of a person infected with coronavirus. Covid-19 spectrum also varies from asymptomatic forms to respiratory failure that needs mechanical ventilation in ICU.2
Viral infection produces excessive immune reaction in the host labelled as cytokines Storm.In SARS CoV-2,viral protein structures like main protease enzyme (3CL pro),viral papain like protease,RNA dependant RNA polymerase (RdRp),helicase enzyme,spike protein could be targeted for conducting homology modelling for discovery of novel drugs against coronavirus.3 Non structural protein nsp 12, which is RNA dependant RNA polymerase is important in replication and transcription of corona vial genome.4 Till now no drugs or vaccine has been approved by regulatory agencies for the treatment of covid-19. But drugs Chloroquine and its analogue hydroxychloroquine has been tried in treatment of corona virus in some countries.
Chloroquine was found to inhibit the growth of SARS-CoV 2 in vitro. Chloroquine rise the pH of endosomal vesicles inside the cells that are points of entry by virus, thereby preventing fusion and entry of virus. It also controls the cytokine storm in the late phase of critically ill covid-19 patients.5
Quinine which is obtained from cinchona bark also used in the treatment of malaria. It has been proved in invitro study that quinine sulphate reduces the multiplication and adsorption of herpes virus.6 Doxycycline,which is second generation tetracycline used in the treatment of severe bacterial infection also proved its effect in invitro studies against dengue virus replication and also against vesicular stomatitis.In a dose dependant manner, Doxycycline inhibited vesicular stomatitis virue replication at concentrations of 1.0–2.0 μg ml.7 So, as a method of drug repurposing,these drugs were chosen to analyse its effect against coronavirus by insilico method.
To speed up the drug designing process, rational drug design method was developed. Docking of drug molecule with receptor or enzyme is one such method.In this study ,effort has been made to design drugs for covid-19 using doxy cycline, quinine by analysing its structure activity relationship and also docking these drugs against Corona virus target protein which in study is non structural protein (nsp 12).
Materials and Methods
Sequence Retrieval
The nucleotide sequence of severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome (ID: NC_045512.2) was retrieved from GenBank nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore/nc_045512.2). The EMBOSS transeq was used to translate the DNA sequence to the corresponding amino acid sequence.8
Template Selection
The amino acid sequence was submitted to BLASTp against protein data bank (PDB) to identify the structurally homologous proteins.9 From the BLAST result the template with maximum identity were aligned with the target sequence using MultAlin, online software for multiple sequence alignment.10
Protein Structure Prediction and Validation
The three dimensional structure of the target protein was determined using SWISS-MODEL (https://swissmodel.expasy.org/assess). SWISS-MODEL is a fully automated server that works based on the principles of homology modeling.11 The modeled structure was energy minimized using Swiss PDB Viewer, an application that reduces the energy of the protein by repairing bond length, bond angles and other distorted geometries of the protein.12 The quality of the predicted model was analyzed using PROCHEK Ramachandran plot and ERRAT.13,14
Analysis of Physico-Chemical Characteristics
The protein’s molecular weight (MW), extinction coefficient, isoelectric point (pI), number of negative and positive charge amino acids (-R, +R), aliphatic index (AI), Grand Average Hydropathy (GRAVY), instability index (II) was determined using Prot Param server.15
N-glycosylation site Prediction
Glycosylation sites in the target viral protein were determined using the web servers GLYCOPP and N-Glycosite, online servers for prokaryotes glycosites prediction .16,17
Secondary Structure and Active Site Prediction
The secondary structure of the protein was determined using self-optimized prediction method (SOPMA)18. Active site in the protein was determined using CASTp. The active site of the amino acids is required for the further molecular docking studies.19 Molecular docking analysis was performed to identify the interaction nature of ligand molecules. Auto Dock is an automatic docking tool and it is designed to predict how small molecules, such as substrates, inhibitors are bind to a receptor of known 3D protein structures. A graphical user interface called Auto Dock Tools or ADT was utilized to generate grids, calculate the dock score. The software identifies the binding interactions of the reference molecule and the extracted molecule to the target protein molecule of the interest, Initially the molecule were read in the autodock software and the grid parameter file was formed and commanded to run autogrid and autodock. Once the dlg files of the molecules were performed by the software, which contains the docking score of the interaction, the file was opened in the Discovery studio for the ligand interaction.
Discovery Studio Visualizer 3.1
A widely available free software that helps in envisaging the macromolecule-ligand interactions, Discovery Studio Visualizer 3.1, developed and distributed by Accelrys, is used to study the simulation of small molecules and large macromolecules. This module helps in determining the structure based design, ligand interaction with the protein, validation and optimization. This also helps to analyze the structure activity relationship between protein and the ligand bind to it.
Results and Discussion
Generation of 3D Structure and Validation
The experimental structure of severe acute respiratory syndrome coronavirus 2 is not determined using X-RAY crystallography and NMR methods. Hence in silico based techniques were used to predict the 3D structure of the target protein.The target protein,Non structural protein (nsp 12) which is RNA dependant RNA polymerase (RdRp) is important in replication and transcription of corona viral genome.The target protein’s structurally homologous templates were retrieved using BLASTp. From BLAST result, two proteins (PDB ID: 6NUR and 6JYT) that showed maximum identity were chosen. Both template sequences were aligned with the target sequence using MultAlin. Finally, the protein 6NUR that showed maximum alignment was chosen as the template. SWISS MODEL, free online homology modeling software was used to predict the structure of the target protein from the structure of template protein. The predicted protein structure was energy minimized using Swiss PDB Viewer. Energy minimization is an important step in the optimization of geometry of the predicted structure. Finally the structure of the protein was visualized using Pymol molecular graphics system.20 Fig 1 shows the energy minimized structure of target protein.
 |
Figure 1: Structure of severe acute respiratory syndrome coronavirus 2 target protein |
The validation of the protein structure was performed using PROCHECK Ramachandran plot and ERRAT. The Ramachandran plot is the plot of phi (φ) and psi (ψ) angles. The result showed that the number of amino acids in the disallowed region is zero and residues in the most favored region are 92.5%. A good quality model will have >90% amino acid in the most favored region.21 The quality factor of the protein from ERRAT was 95.9157%. The quality factor >50% is considered to be a good quality model.22 Fig 2 and Fig 3shows the Ramachandran plot and ERRAT result of the protein respectively.
 |
Figure 2: Ramachandran Plot determined by PROCHECK. The regions in the red indicates favoured region, yellow shows allowed region, light yellow indicate generously allowed region of amino acid and white region indicate disallowed region. |
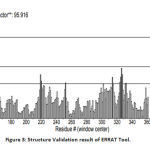 |
Figure 3: Structure Validation result of ERRAT Tool. |
Physico-Chemical Characterization
The physical and chemical properties of the protein was analyzed using Prot Param server, an online server for analysis of molecular weight, isoelectric point and many other properties of the protein. The half life of the protein in vitro mammalian reticulocytes is 0.8 hours. As proteins absorb well at 280nM, the extinction coefficient is calculated at this wavelength. The aliphatic index determines the thermostability of the protein. The results suggest that the protein is heat stable. The Grand Average hydropathy (GRAVY) indices are used to determine the hydrophobicity of the protein. It is measured by dividing the sum of hydropathy values of all the amino acids to the total number of residues in the protein. A GRAVY score below 0 is considered as hydrophilic globular protein.23 Instability index value determines the stability of the protein. A protein with instability index less than 40 is considered stable .24 Table 1 shows the results of physico-chemical properties of the protein.
Table 1: Physico-chemical Characteristics of target protein
| MW (Da) | -R | +R | pI | EC (M-1cm-1) | AI | GRAVY | II |
| 91839.54 | 89 | 79 | 6.14 | 126990 | 80.35 | -0.202 | 27.96 |
N-glycosylation site Prediction
Glycosylation is the important post translational modification in prokaryotic protein, and in many cases it is essential for its binding to the host cell .25 The results from N-Glycosite showed that the protein has two potential N-glycosylation sites. The result was further confirmed using GLYCOPP that also showed two glycosylation sites.
Secondary Structure Prediction
Secondary structure is the folded structure of protein that is formed due to the backbone atoms interaction with each other. The common secondary structures are alpha helix, beta sheets and coils. The secondary structure prediction is important to study the function and activity of the protein.26 The secondary structure of the protein was determined using SOPMA, default parameters of window width 17, similarity threshold 8, and number of states 4 were used for the study. The result of secondary structure in the target sequence is shown in Table 2.
Table 2: Secondary structure elements in target protein
| Secondary Structure | % |
| Alpha helix | 41.97 |
| 310 helix | 0.00 |
| Pi helix | 0.00 |
| Beta bridge | 0.00 |
| Extended strand | 16.94 |
| Beta turn | 9.84 |
| Bend region | 0.00 |
| Random Coil | 31.26 |
| Ambiguous state | 0.00 |
| Other States | 0.00 |
Active Site Prediction
For setting the grid in docking studies, the determination of active site amino acids of the target protein is important.27The active site residues of the protein were determined using CASTp Server. Table 3 and Fig 5 show CASTp results of the amino acids that are in the active site region.
Table 3: CASTp result of active site amino acids
| ASP | 166 | ASN | 509 | ALA | 560 | GLY | 618 | ASP | 686 |
| VAL | 168 | LYS | 513 | GLY | 561 | TRP | 619 | ALA | 687 |
| GLU | 169 | THR | 542 | VAL | 562 | ASP | 620 | THR | 689 |
| LYS | 413 | GLN | 543 | THR | 567 | TYR | 621 | ALA | 690 |
| LYS | 440 | MET | 544 | ARG | 571 | PRO | 622 | TYR | 691 |
| HIS | 441 | ASN | 545 | GLN | 575 | LYS | 623 | ASN | 693 |
| PHE | 443 | LEU | 546 | LEU | 578 | LYS | 623 | LEU | 760 |
| ASP | 454 | LYS | 547 | LYS | 579 | CYS | 624 | SER | 761 |
| TYR | 457 | TYR | 548 | ALA | 582 | ASP | 625 | ASP | 762 |
| TYR | 458 | ALA | 549 | VAL | 590 | ARG | 626 | ASP | 763 |
| ILE | 496 | ILE | 550 | VAL | 590 | MET | 628 | ALA | 764 |
| VAL | 497 | SER | 551 | ILE | 591 | GLU | 667 | PHE | 795 |
| ASN | 498 | ALA | 552 | GLY | 592 | VAL | 669 | ALA | 799 |
| ASN | 499 | LYS | 553 | THR | 593 | LYS | 678 | LYS | 800 |
| LEU | 500 | ARG | 555 | SER | 594 | THR | 682 | CYS | 801 |
| ASP | 501 | ALA | 556 | LYS | 595 | THR | 682 | TRP | 802 |
| LYS | 502 | ARG | 557 | PHE | 596 | SER | 683 | ||
| SER | 503 | THR | 558 | TRP | 600 | SER | 684 | ||
| GLY | 505 | VAL | 559 | MET | 603 | GLY | 685 |
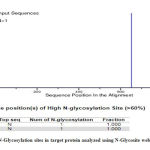 |
Figure 4: N-Glycosylation sites in target protein analyzed using N-Glycosite web server |
 |
Figure 5: CASTp result of active site amino acid residues |
Structure activity relationship and Docking result of Doxycycline against corona target protein (nsp 12)
The structure activity relationship for the Doxycycline compound and the modelled protein revealed that the Doxycycline is well engaged into the active site of the target protein. The compound has strong hydrogen bonding interaction with the non-polar amino acids. The compound(Doxycycline) also has positively charged interaction with the amino acid Arg557, Arg626 and Lys800. The bulkier phenolic group present in the compound makes the compound engage into the active site of the protein more effectively. This also leads to stacking interaction with positively charged aminoacid Arg555 and Arg557. The docking score of the compound(Doxycycline) is found to -7.34 kcal/mol as shown in Table 4. The compound also shows vander wals force interaction with the aminoacid Asp620 and Asp762. The non-structural protein have stacked the compound inside its active site which helps in targeting the polymerases effectively by the compound doxycycline. This will terminate the replication of RNA which further stops the transcription and production of viral proteins. Further targeting the non-structural protein with the drug shows competitive inhibition towards the drug doxycycline. The binding analysis and the ligand interaction is depicted in the Figure.6.
Table 4: Docking score and the interaction for the ligand compounds
| Compound | Binding energy kcal/mol | Atoms forming Hydrogen bond | Other interactions |
| Quinine | -6.14 | ASP 625, ASN 693 | SER 684, THR 682, ASP 762, CYS 624, LYS 623, TYR 621, ASP 620, PRO 622, LYS 800 |
| Doxycycline | -7.34 | ARG 555, ASP 620, CYS 624, ASP 625 | ARG 557, THR 558, PRO 622, TYR 621, LYS 623, ARG 626, ASP 762, LYS 800 |
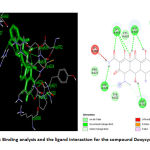 |
Figure 6: Binding analysis and the ligand interaction for the compound Doxycycline |
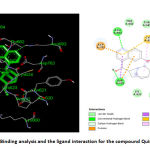 |
Figure 7: Binding analysis and the ligand interaction for the compound Quinine |
Structure Activity Relationship and Docking Result of Quinine Against Corona Target Protein (nsp 12)
The binding analysis and the ligand interaction for the compound (Quinine) revealed that the compound is well interacted with hydrogen bonding interaction with Asn693 and Asp635, negatively charged amino acid Asp762 and the Pi-Pi interaction with Cys624. The compound Quinine is slightly away from the active site due to anionic interaction with Asp762. The carbonyl group in the benzene ring is away from the active site of the Polymerase protein. The compound is also less surrounded by the non-polar aminoacids like Pro622, Tyr621 etc. This makes this compound less potent than the Doxycline. The compound Quinine also doesn’t possess any vander wals interaction with the aminoacids present in the modelled protein Polymerases. Vander wals forces plays an important role in Protein-Ligand binding. Thus failing of Vanderwals force may lead this compound energy level lesser than doxycycline. Thus the docking score of the compound Quinine was found to be -6.14 kcal/mol as shown in Table 4.The binding analysis and the ligand interaction for the compound quinine is depicted in the figure. 7.
Conclusion
The Compounds Doxycycline and Quinine found to have good binding affinity with corona viral non structural protein. Predicting the structure activity relationship of both drugs has also shown that,they have well engaged in the active site of the protein with hydrogen bond interaction. This can lead to novel drug identification of anti-viral therapy towards Covid-19. As this is the current pandemic, this can be used as a initial lead identification. Further, the current results need to be validated by in-vitro and in-vivo anti-viral method for further medicinal usage in corona virus treatment.
Acknowledgement
I thank faculty members of Biotechology department of Vels University,chennai for their contribution in doing docking study in this study .
Conflict of Interest
There is no conflict of interest.
Funding Source
There is no funding source.
References
- Adhikari SP, Meng S, Wu YJ, et al. Epidemiology, causes, clinical manifestation and diagnosis, prevention and control of coronavirus disease (COVID-19) during the early outbreak period: A scoping review. Infect Dis Poverty, 2020;9(29):1-12.
CrossRef - Guan W, Ni Z, Hu Y, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med,2020:1-13.
- Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B, 2020:1-44.
CrossRef - EJ Snijder et al.The nonstructural proteins directing corona virus RNA synthesis and processing. Adv Virus Res,2016;96:59-126.
CrossRef - Wang M,Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res, 2020 ;30: 269–271.
CrossRef - Wolf R, Baroni A, Greco R, et al. Quinine sulfate and HSV replication. Dermatol Online J, 2003; 9(3): 3
CrossRef - Zhuan-chang Wu , Xin Wang , Jian-chao Wei, Bei-bei Li, Dong-hua Shao, et al.Antiviral activity of doxycycline against vesicular stomatitis virus in vitro.FEMS Microbio lett, 2015;362(22)
CrossRef - Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey AR, Potter SC, Finn RD, Lopez R. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic acids Res., 2019;47:636-41.
CrossRef - Garba L, Yussoff MA, Halim KB, Ishak SN, Ali MS, Oslan SN, Rahman RN. Homology modeling and docking studies of a Δ9-fatty acid desaturase from a Cold-tolerant Pseudomonas sp. AMS8. PeerJ, 2018; 6:e4347.
CrossRef - Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic acids Res., 1988;16(22):10881-90.
CrossRef - Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TA, Rempfer C, Bordoli L, Lepore R. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids Res., 2018 ;46(W1):W296-303.
CrossRef - Guex N, Peitsch MC. SWISS‐MODEL and the Swiss‐Pdb Viewer: an environment for comparative protein modeling. electrophoresis,1997;18(15):2714-23.
CrossRef - Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr.,1993; 26(2):283-91.
CrossRef - Messaoudi A, Belguith H, Hamida JB. Homology modeling and virtual screening approaches to identify potent inhibitors of VEB-1 β-lactamase. Theor. Biol. Medl Modell., 2013;10(1):22.
CrossRef - Garg VK, Avashthi H, Tiwari A, Jain PA, Ramkete PW, Kayastha AM, Singh VK. MFPPI–Multi FASTA ProtParam Interface. Bioinformation, 2016;12(2):74.
CrossRef - Chauhan JS, Bhat AH, Raghava GP, Rao A. GlycoPP: a webserver for prediction of N-and O-glycosites in prokaryotic protein sequences. PloS one,2012;7(7):1-12
CrossRef - Zhang M, Gaschen B, Blay W, Foley B, Haigwood N, Kuiken C, Korber B. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology, 2004 ;14(12):1229-46.
CrossRef - Geourjon C, Deleage G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci,1995;11:681–4
CrossRef - Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated resiudes. Nucleic Acids Res, 2006;34(Web Server issue):W116–8.
CrossRef - Lua RC, Lichtarge O. PyETV: a PyMOL evolutionary trace viewer to analyze functional site predictions in protein complexes. Bioinformatics, 2010 ;26(23):2981-2.
CrossRef - Rai DK, Rieder E. Homology modeling and analysis of structure predictions of the bovine rhinitis B virus RNA dependent RNA polymerase (RdRp). Int. J. Mol. sci., 2012 ;13(7):8998-9013.
CrossRef - Elengoe A, Naser MA, Hamdan S. Modeling and docking studies on novel mutants (K71L and T204V) of the ATPase domain of human heat shock 70 kDa protein 1.Int. J. Mol. sci. ,2014 ;15(4):6797-814.
CrossRef - Magdeldin S, Yoshida Y, Li H, Maeda Y, Yokoyama M, Enany S, Zhang Y, Xu B, Fujinaka H, Yaoita E, Sasaki S. Murine colon proteome and characterization of the protein pathways. BioData Min., 2012 ;5(1):11.
CrossRef - Satyanarayana SD, Krishna MS, Kumar PP, Jeereddy S. In silico structural homology modeling of nif A protein of rhizobial strains in selective legume plants. J. Genet. Eng. Biotechnol., 2018 ;16(2):731-7.
CrossRef - Parsons LM, Bouwman KM, Azurmendi H, de Vries RP, Cipollo JF, Verheije MH. Glycosylation of the viral attachment protein of avian coronavirus is essential for host cell and receptor binding. J. Biol. Chem., 2019 ;294(19):7797-809.
CrossRef - Rehman I, Botelho S. Biochemistry, secondary protein structure. InStatPearls [Internet] 2019 Jun 3. StatPearls Publishing.
CrossRef - Ullah M, Ghosh T, Ishaque N, Absar N, Hira J. A Bioinformatics Approach for Homology Modeling Binding Site Identification of Triosephosphate Isomerase from Plasmodium falciparum 3D7. J. Young Pharm., 2012 ;4(4):261-6.
CrossRef

