
Padmashree A.P , Sabia Imran, Tejashree Prakash and Lokesh Ravi*
, Sabia Imran, Tejashree Prakash and Lokesh Ravi*
1Department of Botany, St. Joseph’s College (Autonomous), Bengaluru-27, Karnataka, India.
Corresponding author E-mail: lokesh.ravi@sjc.ac.in
DOI : https://dx.doi.org/10.13005/bpj/2024
Abstract
Aim of this study is to prepare a dataset of 3D protein structures by homology modeling for a fungal pathogen Erysiphe necator that causes “Powdery Mildew” disease in the grapevine crop, Vitis vinifera species commonly known as grape. To construct a 3D structures of protein drug targets, databases such as UniProt KB, Drug Bank, PMDB and online tools such as BLASTp, SWISSModel, Ramachandran plot were used. Total of 100 proteins were selected from E.necator and were screened for potential drug targets. Among these 66 protein were identified as drug targets. These selected proteins were subjected for BLASTp to identify suitable templates for homology modeling. These 66 proteins were subjected for homology modeling construction via SWISS model webtool. Further the inbuilt ramachandran plot analysis in Swiss model website was used to screen the quality of the constructed homology models. Computational structures with reliable quality in the ramachandran plot analysis are then submitted to PMDB online database. Further to understand the application of the constructed homology models, these structures were employed in protein-ligand docking study using tebuconazole and carboxin antibiotics against their drug targets. Among these two antibiotics, tebuconazole was identified to be a potential antifungal that could be employed in control of E.necator pathogen. Further, these constructed models could be emoployed in computational drug discovery and drug development, targeting the E.necator fungus. Thus helping the grape cultivation and improving economic returns from grape and wine production.
Keywords
AutoDock; Erysiphe necator; Homology modeling; Protein drug targets; PMDB.
Download this article as:| Copy the following to cite this article: Padmashree A. P, Imran S, Prakash T, Ravi L. Construction of 3D Model of Protein Drug Targets for Erysiphe Necator a Fungal Plant Pathogen Causing Powdery Mildew. Biomed Pharmacol J 2020;13(3). |
| Copy the following to cite this URL: Padmashree A. P, Imran S, Prakash T, Ravi L. Construction of 3D Model of Protein Drug Targets for Erysiphe Necator a Fungal Plant Pathogen Causing Powdery Mildew. Biomed Pharmacol J 2020;13(3). Available from: https://bit.ly/3nYgskE |
Introduction
Grape is one of the most important economical and commercial fruit for the world and it is a major contribution to the country’s GDP (Gross domestic product) and it is a wide adaptability crop1,2 Vitis vinifera is a dicotyledonous and annual crop plant species that is a member of the Vitaceae family, commonly called “grape” and “draksha”3 it’s cultivated in temperate, sub-tropical, tropical regions, all over the world.3,1,4 Global production of grape is estimated to 67 million metric tons per annum at present. China is one of the leading country in the production of grape with 8,651.83 thousand tons, followed by Italy (7,787.83 thousand tons), the united states of America (6,777.73 thousand tons), Spain (6,107.20 thousand tons), France, Turkey, Argentina, chile, and south Africa.1 India occupies the eighteenth position in world for production of grape and it’s cultivated in an area of 111.4 thousand hectares with a production of 1,234.9 thousand tons.1 Almost 71% of global grape production is used for wine, 27% fresh fruit, 2% dried fruit.3 India’s, 90% of the grape is used for wine production, the rest of the grape is used for fresh eating purpose.1 A total of 60% of the world’s grapevine consumers are Europe, Italy, Spain, France, Turkey, Argentina, Chile, South Africa and are also the dominating world’s wine producers.5,1 Wine caliber depends greatly on the grape quality, with need to procure healthy and quality grape, the cultivar must take gentle care in the prevention of pathogen (fungus) attack on the grapevine.2 The most disastrous disease of grape family is the powdery mildew disease, caused by the ascomycetous pathogen (fungus) Erysiphe necator infects all green tissues of the Vitaceae family.3,6,7 Worldwide grape cultivars are greatly affected by this powdery mildew disease, that decreased there production, economic growth and affected countries such as China, USA, Australia, Italy, Spain, France, Turkey, Argentina, Chile, South Africa.1
Grapevine powdery mildew disease which is caused by the obligately biotrophic pathogen Erysiphe necator (syn. Uncinula necator) that causes the occurrence of white powder mildew disease.8,6,9 This disastrous disease believed to be originated in wild North America in 1845, from there it spread to Europe in the mid-1850s and Australia in 1866.1,5 The fungus develops as superficial hyphae in an epidermal cell of green tissue.5 All green tissues of the Vitaceae family are infected by this fungus, i.e., berries, pedicels, shoots, leaves, and buds.4,9
During winter mycelia and conidia (asexual stage) are present in dormant buds, these are activated at bud burst and the infected flag shoots are covered with mycelia and conidia.10 Conidia are wind-dispersed, thus it initiates multiple infections on green tissues throughout the season. During late summer cleistothecia (sexual stage) are produced on the leaves, shoots, berries and it develops into cleistothecium.9 Ascus contain ascospores, when precipitation coincides with a temperature above 10ºC ascospores are released and starts to infect leaves and berries forms powdery mildew colonies.2,6,7
Symptoms of powdery mildew:
Irregular chlorosis of green tissues becoming grey-white with white powder on the upper and lower surface of the leaf.10
Black net lines with white powder on berry stalk and tendril surface.5
Powdery mildew decreases the development of grapes and causes berry crack, resulting in loss of berry quality and grape production.8
Crop yield decreases with an increase in acidity and decreases anthocyanin and decreases sugar content of mature grapefruit.2,5
Powdery mildew diseases greatly affect yield and economic profit.1
In this study, the 3D structures of proteins of Erysiphe necator, are developed using homology modelling technique.11,12,13,14 These structures are necessary to design and develop drugs that could aim to stop the spread of this dreadful disease.10 Lack of protein structures has hindered the understanding of binding specificities of proteins and ligands, which are pre-requisites for drug design and development.15 Methods of homology modelling are employed to develop protein structures of Erysiphe necator. Homology modelling works on the commonly known fact that proteins with similar sequences have similar structures.12,16 There are varieties of tools available that assist users in homology modelling, which are accessible as downloadable software or online tools.17,14
Materials and Methods
This NCBI database was primarily used to search and identify the organism (https://www.ncbi.nlm.nih.gov/).12 Agricultural pathogens affecting crop plants were screened in the NCBI database to identify the under-explored pathogens to subject for this current study. The protein sequences of identified pathogens were retrieved from Uniprot (Universal Protein knowledge Base: www.uniprot.org).18,19 The sequences were examined and downloaded in .fasta file format.17,18 Protein Basic Local Alignment Search Tool (pBLAST: https://blast.ncbi.nlm.nih.gov/) was used to identify template structures with greater than 80% sequence similarity for homology modelling in the PDB database (Protein Data Bank: www.rcsb.org).15 SWISS Model (https://swissmodel.expasy.org/) was used to construct 3D structures of the selected protein sequences.13,16,14 Based on the selected homology templates, the query sequences were subjected for construction of computational protein models.20 Ramachandran plot analysis inbuilt within the Swiss Model website was used to select the best model among the multiple constructed models for each protein drug target.21,12 Protein Model DataBase (PMDB: https://bioinformatics.cineca.it/PMDB/) is a public database for computational protein models that can be accessed by researchers to access quality protein structures generated computationally.22,20 AutoDock 4.2 was used to perform protein-ligand docking analysis the interactions between protein and ligand are examined using PyMOL tool.16,23,24
Results and Discussion
Drugability Protein Selection
A total of 100 proteins of Erysiphe necator were selected from the UniProtKB database. These proteins were examined in drug bank to find a match to pre-existing reported drug targets. Among the 100 selected protein sequences, 66 proteins were identified as potential drug targets for further processing.
Erecting Homology Model
Amino acid sequences of the selected 66 proteins were retrieved from the UniProt KB database as fasta file format (.fasta). These sequences were subjected for pblast analysis to find a match with entries in PDB (Protein Data Base: www.rcsb.org) to identify protein structures with greater than 80% sequence similarity to select a template for homology modeling. All 66 protein sequences had significant sequence similarity with more than 80% match with existing protein entries in PDB website. Using these identified similarity structures, the 66 protein sequences were subjected to computational homology model construction. Homology models were constructed using the online web tool SWISSModel (https://swissmodel.expasy.org/). The webtool constructed multiple models for each protein. Among the constructed protein models, the best one was selected using Ramachandran plot analysis.
Ramachandran Plot Analysis
The ramachandran plot analysis was performed for all protein model structures constructed in SWISSModel tool. The protein models that demonstrated more than 90% of residues in the favored regions of the ramachandran plot were considered to be reliable for structural applications. All 66 proteins demonstrated above 90% of the residues within the favored regions in the ramachandran plot and hence were considered for further processing. Graphical representation of ramachandran plot analysis of a preferred protein model with 100% of residues in favored region and a least preferred model with 82% of residues within the favored region are shown in Figure.1.
PMDB ID Submission
All 66 protein model structures that were verified using ramachandran plot were then submitted to PMDB (Protein Model Data Base: srv00.recas.ba.infn.it › PMDB) website. The protein model structures were submitted to public database for easy access to researchers to further applications of the same. The list of constructed protein models that were submitted to PMDB is tabulated in Table.1.
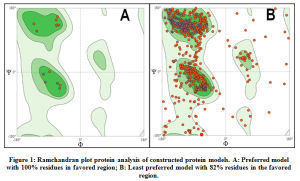 |
Figure 1: Ramchandran plot protein analysis of constructed protein models. A: Preferred model with 100% residues in favored region; B: Least preferred model with 82% residues in the favored region. |
Table.1: Modeled protein structure submissions to PMDB
| Sl. No. | Drug target name | UniProt ID | Confident Score | PMDB
ID |
| 1 | ATP-dependent DNA helicase PIF1 | A0A0B1PEI0 | 93.17% | PM0082721 |
| 2 | Adenylyltransferase and sulfurtransferase uba4 | A0A0B1P610 | 92.91% | PM0082722 |
| 3 | Phosphatidylserine decarboxylase proenzyme 2 | A0A0B1P526 | 93.55% | PM0082728 |
| 4 | Putative myosin class v myosin | A0A0B1P6S1 | 93.78% | PM0082816 |
| 5 | ATP-dependent 6-phosphofructokinase | A0A0B1PDE5 | 93.77% | PM0082731 |
| 6 | NADPH–cytochrome P450 reductase | A0A0B1P387 | 95.47% | PM0082732 |
| 7 | Proliferating cell nuclear antigen | A0A0B1NZA1 | 96.71% | PM0082733 |
| 8 | Adenylate kinase | A0A0B1P0M5 | 95.79% | PM0082735 |
| 9 | GTP:AMP Phosphotransferase | A0A0B1PEI1 | 90.66% | PM0082736 |
| 10 | Glutathione reductase | A0A0B1P915 | 94.69% | PM0082738 |
| 11 | Arginine biosynthesis bifunctional protein Argl | A0A0B1P8H8 | 92.82% | PM0082741 |
| 12 | Eburicol 14-alpha-demethylase | O14442 | 96.39% | PM0082742 |
| 13 | Inositol hexakisphosphate and diphosphoinositol-pentakisphosphate kinase | A0A0B1P4A3 | 93.26% | PM0082743 |
| 14 | Endonuclease III homolog | A0A0B1NYA2 | 91.42% | PM0082744 |
| 15 | Ubiquinone biosynthesis O-methyltransferase | A0A0B1P3U8 | 91.67% | PM0082746 |
| 16 | Uridylate kinase | A0A0B1PFZ3 | 96.81% | PM0082747 |
| 17 | DNA repair protein RAD51 homolog | A0A0B1PBG3 | 92.60% | PM0082748 |
| 18 | NADPH-dependent diflavinoxidoreductase 1 | A0A0B1PIW3 | 91.00% | PM0082749 |
| 19 | Phosphatidyl-N-methylethanolamine N-methyltransferase | A0A0B1P4Q8 | 97.96% | PM0082750 |
| 20 | Adenylosuccinatesynthetase | A0A0B1PBI9 | 93.32% | PM0082752 |
| 21 | QueuinetRNA-ribosyltransferase catalytic subunit 1 | A0A0B1NYU0 | 93.33% | PM0082755 |
| 22 | Non-specific serine/threonine 23protein kinase | A0A0B1P860 | 91.15% | PM0082756 |
| 23 | Multifunctional tryptophan biosynthesis protein | A0A0B1P0S5 | 94.68% | PM0082757 |
| 24 | Succinate–CoA ligase [ADP-forming] subunit beta | A0A0B1P9X5 | 96.54% | PM0082758 |
| 25 | Tubulin gamma chain | A0A0B1P6M2 | 95.82% | PM0082759 |
| 26 | Tubulin beta chain (Beta-tubulin) | Q86ZP5 | 97.88% | PM0082761 |
| 27 | Tubulin beta chain | A0A0B1NYP5 | 97.64% | PM0082763 |
| 28 | Catalase-peroxidase | A0A0B1P921 | 95.32% | PM0082764 |
| 29 | tRNA (guanine-N(7)-)-methyltransferase | A0A0B1P4Q1 | 93.52% | PM0082765 |
| 30 | Double-strand break repair protein | A0A0B1PBX4 | 100.00% | PM0082766 |
| 31 | Non-specific serine/threonine protein kinase | A0A0B1PCS0 | 93.24% | PM0082767 |
| 32 | Ketol-acid reductoisomerase | A0A0B1P276 | 93.32% | PM0082768 |
| 33 | Replication protein A subunit | A0A0B1PG76 | 92.11% | PM0082769 |
| 34 | Alanine –tRNA ligase | A0A0B1PHH9 | 96.53% | PM0082770 |
| 35 | Methionine aminopeptidase 2 | A0A0B1PC83 | 94.85% | PM0082771 |
| 36 | tRNA N6-adenosine threonylcarbamoyltransferase | A0A0B1P9F1 | 90.27% | PM0082772 |
| 37 | CDP-diacylglycerol- -serine phosphatidyltransferase | A0A0B1P6T8 | 94.09% | PM0082773 |
| 38 | Serine/threonine-protein phosphatase 2A 56 kDa regulatory subunit | A0A0B1PJ13 | 95.98% | PM0082774 |
| 39 | Eukaryotic translation initiation factor 6 | A0A0B1P4I8 | 95.95% | PM0082777 |
| 40 | Serine/threonine-protein kinase Tel1 | A0A0B1P515 | 92.40% | PM0082778 |
| 41 | tRNA (guanine(37)-N1)-methyltransferase | A0A0B1NYC9 | 96.46% | PM0082779 |
| 42 | Imidazole glycerol phosphate synthase hisHF | A0A0B1PC64 | 95.54% | PM0082780 |
| 43 | Succinate—CoA ligase[ADP-forming] subunit alpha | A0A0B1P7X3 | 97.33% | PM0082782 |
| 44 | Kinesin-like protein | A0A0B1P242 | 91.48% | PM0082783 |
| 45 | NAD(P)H-hydrate epimerase | A0A0B1P5V7 | 92.48% | PM0082784 |
| 46 | 1,2-dihydroxy -3-keto-methylthiopentene-deoxygenase | A0A0B1PEY8 | 95.29% | PM0082785 |
| 47 | Phosphoacetylglucosamine mutase | A0A0B1PEY4 | 94.31% | PM0082787 |
| 48 | 2-methoxy-6-polyprenyl-1,4-benzoquinol methylase | A0A0B1NVN7 | 93.48% | PM0082789 |
| 49 | Deoxyhypusine hydroxylase | A0A0B1P017 | 94.86%
|
PM0082790
|
| 50 | RuvB-like helicase | A0A0B1PC66 | 97.32% | PM0082791 |
| 51 | Mitogen-activated protein kinase | A0A0B1P7C2 | 95.05% | PM0082792 |
| 52 | Translation factor GUF1 | A0A0B1PC47 | 90.19% | PM0082793 |
| 53 | Methylthioribulose-1-phosphate dehydratase | A0A0B1P0T7 | 93.81% | PM0082794 |
| 54 | Methionine aminopeptidase 2 | A0A0B1PHZ3 | 95.12% | PM0082796 |
| 55 | Methionine aminopeptidase 2 | A0A0B1P0D0 | 94.20% | PM0082799 |
| 56 | Vacuolar proton pump subunit B | A0A0B1NX09 | 92.81% | PM0082800 |
| 57 | 3-hydroxy-3-methylglutaryl coenzyme A reductase | A0A0B1P2R9 | 94.29% | PM0082802 |
| 58 | Ceramide very long chain fatty acid hydroxylase | A0A0B1P141 | 95.07% | PM0082803 |
| 59 | Patatin-like phospholipase domain-containing protein | A0A0B1P5M0 | 98.19% | PM0082804 |
| 60 | mRNA-capping enzyme subunit alpha | A0A0B1P5M6 | 94.12%
|
PM0082805 |
| 61 | Histidine biosynthesis trifunctional protein | A0A0B1P4Z9 | 95.97% | PM0082806 |
| 62 | NADH Dehydrogenase flavoprotein1 | A0A0B1P2X3 | 94.09% | PM0082807 |
| 63 | Succinate dehydrogenase[ubiquinone] iron-sulfur subunit | A0A0B1PFX7 | 90.95% | PM0082810 |
| 64 | Nicotinatephosphoribosyltransferase | A0A0B1PFX6 | 95.32% | PM0082811 |
| 65 | Maintenance of mitochondrial morphology protein 1 | A0A0B1P737 | 95.65% | PM0082812 |
| 66 | Lipoyl synthase | A0A0B1PCV5 | 95.49% | PM0082815 |
Protein-Ligand Docking
The application of these constructed protein models is to involve in structure based computational drug design. To test the SBCADD applications of the protein models, two known antifungal drugs (i.e., tebuconazole and carboxin) were subjected for protein-ligand docking with their reported protein drug targets, that are modeled in this study (i.e., Cytochrome P450 Reductase & Succinate Dehydrogenase) respectively. Results of the docking study suggests that Among the two analyzed antifungal agents tebuconazole demonstrated higher potential to be an effective inhibitor of Cytochrome P450 reductase protein, with a binding energy of -7.6Kcal/mol with formation of 1 hydrogen bond with Asp-691 and hydrophobic interaction with Thr-074, Arg-530, Gln-073, Tyr-451, His-315, Tyr-125, Trp-693, Asp-691, Gly-126, Leu-167, Val-692, Glu-127, Asn- 169, Asp-202 residues. The antifungal agent carboxin demonstrated an insignificant binding energy of 5.2Kcal/mol with formation of 2 hydrogen bonds with Asp-179 and Asn-256 and formed hydrophobic interactions with Glu-183, Lys-176, Tyr-214, Leu-255, Lys-177, Leu-259, Asp-179, Leu-181, Ala-260, Pro-257, Leu-255, Asn-256. The graphical representation of the protein-ligand interactions between the test antibiotics and its protein target are shown in Figure.2. Among the two tested antifungal agent, it could be suggested that Tebuconazole could be an effective drug to control the infection of Erysiphe necator fungus. Thus, the protein models constructed in this study could be employed in similar computational studies, to identify the effective antifungal agents among the existing drugs or can be used to identify new and novel antifungal agents targeting this specific fungal infection.
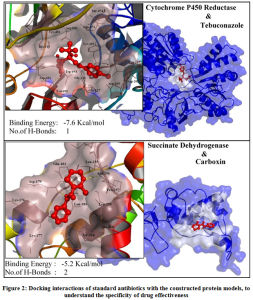 |
Figure 2: Docking interactions of standard antibiotics with the constructed protein models, to understand the specificity of drug effectiveness. |
Conclusion
The current study aimed at construction of 3D computational protein model structures of a un explored fungal pathogen Erysiphe necator4 that causes powdery mildew disease, which causes a great economical impact in grapewine crop cultivation.25 A similar study by Divya et.al., (2018) constructed computational models of protein drug targets of Perkinsus marinus an endoparasitic pathogen that has economical impact in aquaculture of shellfish and mollusks.12 Further to demonstrate the SBCADD application of the constructed protein models, protein-ligand docking analysis was carried between two known antifungal drugs and their drug targets. Among the two test antifungal agents, tebuconazole was found to be a better effective antibiotic that could possibly help control the infection of this pathogen in grape cultivation. However, further in-silico validation and in-vitro studies are required for confirmation.
These protein models that were constructed in this study are made available to the scientific community by submitting to PMDB database. These structures can be exploited in both computational drug discovery and drug development, where existing drugs can be screened with these protein models to identify an effective antibiotic (as demonstrated with an example) further, natural product sources can be screened to identify a potential natural source to control the spread of this infectious fungus. This study opens opportunity for further research in computational drug discovery and design and helps accelerate the in-vitro research on this fungal pathogen.
Acknowledgement
The authors thank the management of St. Joseph’s College (Autonomous), Bengaluru for supporting this research work.
Conflict of Interest
No conflict of interest.
Funding Source
No funding was availed for this work.
References
- Shinde PV, An Economics of Grapes under Horticulture in India. IJRSI, 3(2): 69–71 (2016)
- Pavloušek P, Grapevine breeding in Central and Eastern Europe. Grapevine Breed Programs Wine Ind, 211–244 (2015)
- Reddy CL, BioSciences. RRBS, 7(5): 3–8 (2013)
- Cadle-Davidson L, Chicoine DR, Consolie NH, Variation within and among Vitis spp. for foliar resistance to the powdery mildew pathogen Erysiphe necator. Plant Dis, 95(2): 202–211 (2011)
- Gadoury DM, Cadle-Davidson L, Wilcox WF, Dry IB, Seem RC, Milgroom MG, Grapevine powdery mildew (Erysiphe necator): A fascinating system for the study of the biology, ecology and epidemiology of an obligate biotroph. Mol Plant Pathol, 13(1): 1–16 (2012)
- Gubler WD, Smith RJ, Varela LG, Vasquez S, Stapleton JJ, Purcell AH, Grape Powdery Mildew. NM state Univ, 2013(22 August): 1–4 (2008)
- Qiu W, Feechan A, Dry I, Current understanding of grapevine defense mechanisms against the biotrophic fungus (Erysiphe necator), the causal agent of powdery mildew disease. Hortic Res, 2(April): 1–9 (2015)
- Jones L, Riaz S, Morales-Cruz A, Amrine KCH, McGuire B, Gubler WD, et al., Adaptive genomic structural variation in the grape powdery mildew pathogen, Erysiphe necator. BMC Genomics, 15(1): 1471–2164 (2014)
- Weng K, Li ZQ, Liu RQ, Wang L, Wang YJ, Xu Y, Transcriptome of erysiphe necator-infected vitis pseudoreticulata leaves provides insight into grapevine resistance to powdery mildew. Hortic Res, 1(August): 1–12 (2014)
- Gojiya AU, Pandya JR, Kapadia CV, World’s first report of Erysiphe hyperici causing powdery mildew on fenugreek. Int J Plant Prot, 11(2): 174–176 (2018)
- Satyanarayana SDV, Krishna MSR, Pavan Kumar P, Jeereddy S, In silico structural homology modeling of nif A protein of rhizobial strains in selective legume plants. J Genet Eng Biotechnol, 16(2): 731–737 (2018)
- Jindam D, Ravi L, Krishnan K, Construction of computational protein structure data base by homology modeling for the aquatic pathogen perkinsus marinus for targeted drug design and development. Res J Pharm Technol, 11(6): 2203–2208 (2018)
- Cavasotto CN, Phatak SS, Homology modeling in drug discovery : current trends and applications. 14(July) (2009)
- Joshi YN, Gajul SG, In-silico Homology Modeling of MMP25 involved in Asthma. IJSRT, 4(9): 202–208 (2018)
- Scientific R, International Journal Of. 7(3) (2016)
- Garba L, Mohamad Yussoff MA, Abd Halim KB, Ishak SNH, Mohamad Ali MS, Oslan SN, et al., Homology modeling and docking studies of δ19-fatty acid desaturase from a Cold-tolerant Pseudomonas sp. AMS8. PeerJ, 2018(3): 1–21 (2018)
- Kumar S, Computational identification and binding analysis of orphan human cytochrome P450 4X1 enzyme with substrates. BMC Res Notes, 8(1): 1–10 (2015)
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, et al., UniProt : the Universal Protein knowledgebase. 32 (2004)
- Al-Khayyat MZS, Al-Dabbagh AGA, In silico Prediction and Docking of Tertiary Structure of LuxI, an Inducer Synthase of Vibrio fischeri. Reports Biochem Mol Biol, 4(2): 66–75 (2016)
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al., SWISS-MODEL : modelling protein tertiary and quaternary structure using evolutionary information. 42(April): 252–258 (2014)
- Kumar P, Arya A, Ramachandran Plot : A simplified approach. (December) (2018)
- Arnold K, Kiefer F, Kopp J, Battey JND, Podvinec M, Westbrook JD, et al., The Protein Model Portal. J Struct Funct Genomics, 10(1): 1–8 (2009)
- DeLano WL, The PyMOL Molecular Graphics System, Version 2.0 Schrödinger LLC. (2015)
- Dallakyan S, Olson AJ, Small-molecule library screening by docking with PyRx. Methods Mol Biol, (1263): 243–250 (2015)
- Montarry J, Cartolaro P, Delmotte F, Jolivet J, Willocquet L, Genetic structure and aggressiveness of Erysiphe necator populations during grapevine powdery mildew epidemics. Appl Environ Microbiol, 74(20): 6327–6332 (2008)

