
Natesan Gnanasekaran
Department of Medical Biochemistry, School of Medicine, College of Health Sciences, Addis Ababa University, Ethiopia
Corresponding Author E-mail : ngsbio2@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/1965
Abstract
In this review how the highly active antiretroviral therapy (HAART) damage the mitochondria, followed by immune activation and insulin resistance. The introduction and evolution of HAART over the past 25 years have brought an amazing decrease in AIDS-and immunodeficiency-related reasons for death. However, deaths of these people are now related to metabolic disorder diseases, including atherosclerotic cardiovascular diseases, before from the viral infection itself. Insulin resistance is at the root causes of maladies of metabolic related disorder diseases. While it is notable that HAART metabolism and immune activation cross taking contribute advancement of insulin resistance and co-morbid illnesses. The molecular mechanisms of HAART metabolism and insulin resist is not completely understand. Emerging evidence that HAART how induced mitochondrial damage? and exacerbates the inflammatory response which leads to the development of insulin resistance are remain largely unresolved. The clarification and comprehension of these mechanisms will offer ascent to new classes of medications for the management of insulin resistance that will additionally improve the quality of life of HIV-infected patients.
Keywords
Antiretroviral Therapy (HAART)
Download this article as:| Copy the following to cite this article: Gnanasekaran N. The Missing Link between HAART, Mitochondrial Damage and Insulin Resistance. Biomed Pharmacol J 2020;13(2). |
| Copy the following to cite this URL: Gnanasekaran N. The Missing Link between HAART, Mitochondrial Damage and Insulin Resistance. Biomed Pharmacol J 2020;13(2). Available from: https://bit.ly/2AjbA5C |
Introduction
There were roughly 36.9 million individuals living with HIV towards the end of 2017 with 1.8 million individuals recently infected in 2017 internationally. Among them 21.7 million individuals were under antiretroviral treatment, 59% of adults and 52% of children. There is no remedy for HIV disease. Notwithstanding, viable antiretroviral (ARV) medications can control the infection and help counteract transmission. New HIV positive fell by 36%, and HIV-related deaths fell by 38% with 11.4 million lives spared because of ART in a similar period [1]. Thus, HIV infection is these days considered “only” a chronic disease. There are in 20 affirmed antiretroviral drugs characterized into five groups as per the mechanisms by which they interfere with the HIV life cycle [2]. Longtime utilization of HAART has been identified with drug toxic effect that can bargain non-AIDS illnesses. A portion of these aggravations is insulin resistance followed by metabolic syndrome. This brings hypertriglyceridemia, hypercholesterolemia, hypertension, endocrine disorders, osteopenia , neurocognitive disorders, diabetes, cardiovascular disease, non-AIDS-defining cancers and lipodystrophy have been observed during therapy [3, 4, 5, 6]. Ongoing examinations likewise propose that utilization of HAART, liver disease speaks to a critical reason for morbidity and mortality in HIV-infected patients. These all complication is related to mitochondria damage induced by HAART. The following table listed FAD-approved HIV medication latest update (January 2020)
Table 1: FDA-Approved HIV Medicines
|
Drug |
Generic Name ( other Names and Acronyms
|
Brand name | FAD Approval date |
| 1.Nucleoside Reverse Transcriptase Inhibitors (NRTIs)
|
|||
| NRTIs block reverse transcriptase, an enzyme HIV needs to make copies of itself. | abacavir (abacavir sulfate, ABC) |
Ziagen | December 17, 1998 |
| emtricitabine (FTC) |
Emtriva | July 2, 2003 | |
| lamivudine (3TC) |
Epivir | November 17, 1995 | |
| tenofovir disoproxil fumarate (tenofovir DF, TDF) |
Viread | October 26, 2001 | |
| zidovudine (azidothymidine, AZT, ZDV) |
Retrovir | March 19, 1987 | |
| 2. Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) | |||
| NNRTIs bind to and later alter reverse transcriptase, an enzyme HIV needs to make copies of itself. | doravirine (DOR) |
Pifeltro | August 30, 2018 |
| efavirenz (EFV) |
Sustiva | September 17, 1998 | |
| etravirine (ETR) |
Intelence | January 18, 2008 | |
| nevirapine (extended-release nevirapine, NVP) |
Viramune | June 21, 1996 | |
| Viramune XR (extended release) | March 25, 2011 | ||
| rilpivirine (rilpivirine hydrochloride, RPV) |
Edurant | May 20, 2011 | |
| 3. Protease Inhibitors (PIs) | |||
| PIs block HIV protease, an enzyme HIV needs to make copies of itself. | atazanavir (atazanavir sulfate, ATV) |
Reyataz | June 20, 2003 |
| darunavir (darunavir ethanolate, DRV) |
Prezista | June 23, 2006 | |
| fosamprenavir (fosamprenavir calcium, FOS-APV, FPV) |
Lexiva | October 20, 2003 | |
| ritonavir (RTV)Although ritonavir is a PI, it is generally used as a pharmacokinetic enhancer as recommended in the Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV and the Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection. |
Norvir | March 1, 1996 | |
| saquinavir (saquinavir mesylate, SQV) |
Invirase | December 6, 1995 | |
| tipranavir (TPV) |
Aptivus | June 22, 2005 | |
| 4. Fusion Inhibitors | |||
| Fusion inhibitors block HIV from entering the CD4 cells of the immune system. | enfuvirtide (T-20) |
Fuzeon | March 13, 2003 |
| 5. CCR5 Antagonists | |||
| CCR5 antagonists block CCR5 coreceptors on the surface of certain immune cells that HIV needs to enter the cells. | maraviroc (MVC) |
Selzentry | August 6, 2007 |
| 6.Integrase Inhibitors | |||
| Integrase inhibitors block HIV integrase, an enzyme HIV needs to make copies of itself. | dolutegravir (DTG, dolutegravir sodium) |
Tivicay | August 13, 2013 |
| raltegravir (raltegravir potassium, RAL) |
Isentress | October 12, 2007 | |
| Isentress HD | May 26, 2017 | ||
| 7.Post-Attachment Inhibitors | |||
| Post-attachment inhibitors block CD4 receptors on the surface of certain immune cells that HIV needs to enter the cells. | ibalizumab-uiyk (Hu5A8, IBA, Ibalizumab, TMB-355, TNX-355) |
Trogarzo | March 6, 2018 |
| 8. Pharmacokinetic Enhancers | |||
| Pharmacokinetic enhancers are used in HIV treatment to increase the effectiveness of an HIV medicine included in an HIV regimen. | cobicistat (COBI, c) |
Tybost | September 24, 2014 |
| 9. Combination HIV Medicines | |||
| Combination HIV medicines contain two or more HIV medicines from one or more drug classes. | abacavir and lamivudine (abacavir sulfate / lamivudine, ABC / 3TC) |
Epzicom | August 2, 2004 |
| abacavir, dolutegravir, and lamivudine (abacavir sulfate / dolutegravir sodium / lamivudine, ABC / DTG / 3TC) |
Triumeq | August 22, 2014 | |
| abacavir, lamivudine, and zidovudine (abacavir sulfate / lamivudine / zidovudine, ABC / 3TC / ZDV) |
Trizivir | November 14, 2000 | |
| atazanavir and cobicistat (atazanavir sulfate / cobicistat, ATV / COBI) |
Evotaz | January 29, 2015 | |
| bictegravir, emtricitabine, and tenofovir alafenamide (bictegravir sodium / emtricitabine / tenofovir alafenamide fumarate, BIC / FTC / TAF) |
Biktarvy | February 7, 2018 | |
| darunavir and cobicistat (darunavir ethanolate / cobicistat, DRV / COBI) |
Prezcobix | January 29, 2015 | |
| darunavir, cobicistat, emtricitabine, and tenofovir alafenamide (darunavir ethanolate / cobicistat / emtricitabine / tenofovir AF, darunavir ethanolate / cobicistat / emtricitabine / tenofovir alafenamide, darunavir / cobicistat / emtricitabine / tenofovir AF, darunavir / cobicistat / emtricitabine / tenofovir alafenamide fumarate, DRV / COBI / FTC / TAF) |
Symtuza | July 17, 2018 | |
| dolutegravir and lamivudine (dolutegravir sodium / lamivudine, DTG / 3TC) |
Dovato | April 8, 2019 | |
| dolutegravir and rilpivirine (dolutegravir sodium / rilpivirine hydrochloride, DTG / RPV) |
Juluca | November 21, 2017 | |
| doravirine, lamivudine, and tenofovir disoproxil fumarate (doravirine / lamivudine / TDF, doravirine / lamivudine / tenofovir DF, DOR / 3TC / TDF) |
Delstrigo | August 30, 2018 | |
| efavirenz, emtricitabine, and tenofovir disoproxil fumarate (efavirenz / emtricitabine / tenofovir DF, EFV / FTC / TDF) |
Atripla | July 12, 2006 | |
| efavirenz, lamivudine, and tenofovir disoproxil fumarate (EFV / 3TC / TDF) |
Symfi | March 22, 2018 | |
| efavirenz, lamivudine, and tenofovir disoproxil fumarate (EFV / 3TC / TDF) |
Symfi Lo | February 5, 2018 | |
| elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide (elvitegravir / cobicistat / emtricitabine / tenofovir alafenamide fumarate, EVG / COBI / FTC / TAF) |
Genvoya | November 5, 2015 | |
| elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate (QUAD, EVG / COBI / FTC / TDF) |
Stribild | August 27, 2012 | |
| emtricitabine, rilpivirine, and tenofovir alafenamide (emtricitabine / rilpivirine / tenofovir AF, emtricitabine / rilpivirine / tenofovir alafenamide fumarate, emtricitabine / rilpivirine hydrochloride / tenofovir AF, emtricitabine / rilpivirine hydrochloride / tenofovir alafenamide, emtricitabine / rilpivirine hydrochloride / tenofovir alafenamide fumarate, FTC / RPV / TAF) |
Odefsey | March 1, 2016 | |
| emtricitabine, rilpivirine, and tenofovir disoproxil fumarate (emtricitabine / rilpivirine hydrochloride / tenofovir disoproxil fumarate, emtricitabine / rilpivirine / tenofovir, FTC / RPV / TDF) |
Complera | August 10, 2011 | |
| emtricitabine and tenofovir alafenamide (emtricitabine / tenofovir AF, emtricitabine / tenofovir alafenamide fumarate, FTC / TAF) |
Descovy | April 4, 2016 | |
| emtricitabine and tenofovir disoproxil fumarate (emtricitabine / tenofovir DF, FTC / TDF) |
Truvada | August 2, 2004 | |
| lamivudine and tenofovir disoproxil fumarate (Temixys, 3TC / TDF) |
Cimduo | February 28, 2018 | |
| lamivudine and zidovudine (3TC / ZDV) |
Combivir | September 27, 1997 | |
| lopinavir and ritonavir (ritonavir-boosted lopinavir, LPV/r, LPV / RTV) |
Kaletra | September 15, 2000 | |
HAART
Mitochondrial Dysfunction
Every mammalian cell contains hundred to thousand mitochondria. The size, shape, and number of mitochondria change significantly in various cell types and more may change under various energy requests and distinctive physiological or natural conditions. The principle capacity of mitochondria is to incorporate ATP through electron transport and oxidative phosphorylation in combination with the oxidation of metabolites by a tricarboxylic acid cycle and catabolism of fatty acids by beta-oxidation. In this sense, the mitochondrion viewed as basically the fuel supplier for the most fundamental energy requests of the cells. On the other hand, mitochondria are presently perceived as being basic segments in the control of numerous key cellular processes, being the primary mediator in the inception and execution of apoptosis and important functions in the assurance of life and deaths of the mammalian cells. Also, mitochondria are the principal intracellular source of reactive oxygen species (ROS) [7]. During the electron transport chain in the mitochondria, few electrons “spill” to cytosolic oxygen, forming the superoxide radical [8]. The electron transport complex I of the mitochondria leaks the electron and formation of superoxide. Mitochondria are not only important sources of ROS production, but also the major targets of ROS attack (9). Many things influence the function of mitochondria for example, ageing, infection, and drugs, etc. These factors can injure the mitochondria, affecting the normal functions of the cell. Mitochondrial injury can change the normal functions of heart, nerves, muscles, pancreas, kidneys, and liver, causing many chronic diseases. Mitochondrial damage emerges from a deficient number of mitochondria, a failure to give essential substrates to mitochondria, or an impaired electron transport and ATP-synthesis components. The number and practical status of mitochondria in a cell can be changed by: fusion of partially damaged mitochondria to healthy mitochondria, which renews and improves its functional capacity the formation of new mitochondria by fission and completely damage mitochondria are degraded by mitophagy (10). These occasions are controlled by complex cell processes that sense the damage of mitochondria [11].
HAART and Mitochondria Damage
It is conceivable that chronic infection and inflammation and/ or adverse effect of the impacts of the drug on mitochondrial capacity would add to long-term complications in HIV-infected people. A substantial list of clinical appearances of mitochondrial toxicity has been portrayed within HAART-related adverse events, which is a noteworthy worry for the determination and long-term adherence to a specific treatment. Amid the period of monotherapy with zidovudine (ZDV), a nucleoside reverse transcriptase inhibitor (NRTI), a few patients created skeletal muscle myopathies [12]. Histological examination of their muscle, adipose tissue, heart and liver, biopsies discovered mitochondrial damage [13, 14, 15, 16]. With the broad utilization of NRTIs, other clinical indications, for example, lactic acidosis, lipodystrophy, peripheral neuropathies, cardiomyopathies, and pancytopenia were found [17]. Inhibition of polymerase gamma (Pol- ??), the enzyme involved in the replication of mtDNA, reduces the amount of mitochondrial DNA (mtDNA) content ensuing mitochondrial damage, was embroiled as the hidden system for these toxicities [18]. Reduction of mtDNA has been widely studied in various tissues of human and animal models such as placenta, fetal cord blood, heart, adipose tissue, skeletal muscle, brain and kidney [19,20,21,22,23]. Yet, amassing proof beyond the “Pol-?? speculation” have been brought up in the most recent years proposing that there are different types of mitochondrial damage both related and unrelated to mtDNA [24]. Accordingly, inhibition of mitochondrial RNA expression by NRTI has been seen in numerous cell lines [25] which may happen through mtRNA polymerase inhibition or by the confinement of the essential cofactors for mtRNA transcription. A few NRTIs likewise have a direct inhibitory effect on the function of mitochondria. In this manner, AZT inhibits the mitochondrial adenylate kinase and adenine nucleotide translocator in mitochondria [26]. AZT additionally increases oxidative stress (OS) and applies a direct inhibitory impact on the electron transport chain, resulting in reduction of OXPHOS [27]. NRTIs also initiate a noteworthy decrease in complex IV action through a specific inhibition of complex I [28]. In vivo evaluation with AZT showed a disturbed cardiac mitochondrial ultra structure, decreased expression of cytochrome b mRNA, and increased levels of oxidative stress in mitochondria. Prolonged mitochondrial ROS production has additionally been implicated to go with or even establish a different system of NRTI-initiated mitochondrial harmfulness. Additionally, protease inhibitors (PIs) and nonnucleoside reverse transcriptase inhibitors (NNRTIs) don’t inhibit Pol- gamma, but the cause mitochondrial damage [29, 30]. Hemandez et al reported that pregnant women under HAART medication showed subclinical mitochondrial damage of themselves and their newborn by reduction of mtDNA, mitochondrial protein synthesis and mitochondrial function [31].Various studies reported that mitochondrial dysfunction in peripheral organs results in insulin insensitivity [32, 33, 34, 35] Hence healthy mitochondria and its proper functions are very important for insulin sensitivity.
HAART and Insulin Resistance
Mitochondrial irregularities related with ART have been previously reported. Literature survey shows that HIV itself and ART influence mitochondrial work through a variety of components, incorporating direct increments in ROS, diminished ATP production, and reduced mitochondrial oxygen utilization [36, 37, 38, 39]. Lower mitochondrial respiration is the potential mechanism for IR in people living with HIV (40). A longitudinal investigation of insulin resistance (IR) in individuals living with HIV on recent ART regimens showed IR in 21%, which indicates a huge decline from 35– 63% in IR compared to older ART regimens [41]. PIs of the HAART regimens potential components cause IR [42, 43, 44, 45] even without the changes in body composition [46, 47]. PIs strongly inhibits the insulin-depended glucose transporter Glut 4, prompting to peripheral IR and impaired glucose tolerance. [48].
Molecular Mechanism of Mitochondrial Damage and Insulin Resistance
Pattern recognition receptors (PPRs) are parts of the innate immune framework, whose primary job is detecting the tissue damage, cell stress and infection. The inflammatory reactions get started by NOD- linked receptor (NLR) family of cytoplasmic PRPs. There are 22 known human NLR family members, including NLRP3-inflammasome [49]. Inflammasomes are activated by a variety of physiological and pathogenic signals. Inflammasome activation is a basic segment of the innate immune reaction and is related to the basic clearance of pathogens or damaged cells. The,overall inflammasome activation is likewise a noteworthy driver of autoimmune and metabolic disorders which is fundamental for the understanding of this process in physiological and pathological settings [50].Latest investigations reported that mitochondria are connected with immune responses [51] and non communicable diseases [52, 53, 54]. At the point when mitochondria are injured, the dysfunctional mitochondria raise the level of reactive oxygen species (ROS) in cells and enhance immune response by activating NALP3 inflammasome [55, 56, 57]. Also, current reports suggested that different mitochondrial particles known as damage-associated molecular patterns (DAMPs) for example, mtDNA, cardiolipin, or dynamin-related protein 1 etc. can be dislocated to the exterior of mitochondria (e.g., cytosol, cell surface, or extracellular spaces) and activates the immune response. Different DAMPs can be discharged into cytosol by stressing cells undergoing autophagy or damage without obvious cell deaths. Proof has been brought forth regarding the autophagy-mediated release of high-mobility group protein B1 (HMGB1), ATP, and mtDNA etc. [58]. DAMPs directly activate the inflammasome (Figure 1) , however, ROS initiation of NLRP3 can be done through thioredoxin (TRX). At the point when the cytoplasmic ROS are increased, the reduced TRX gets oxidized and detached from thioredoxin restricting protein (TXNIP) which binds to NLRP3 for activation [59]. NLRP3 inflammasome activation prompts the activation of caspase-1[60]. The activated caspase 1 is converting pronflammatory inactive form of cytokines interleukin-1β (IL-1β) and IL-18 to their active form [61](Figure 1).Recently, the role of the IL-1β pathway in insulin resistance was well described [62, 63, 64]. With regards to the exact fundamental mechanism, an amazing number of genetic animal models presently showed the function of the inflammasome in interceding IL-1β-initiated insulin resistance [64]. IL-1β signaling pathway works not only for insulin resistance but also for defective insulin secretion. It has been accounted for the beta cell failure [65, 66, 67, 68] since the cell itself produces IL-1β upon overload of glucose [65]. This is further supported by an earlier study showing that antagonist IL-1Ra treatment and defect in NALP3, caspase 1 or IL-1β in animal model recovered from hyperglycemia by improving both β cell insulin production and insulin sensitivity in peripheral organs [63, 69].
The utilization of HAART in the course of the last 20 years has changed death rates among people living with HIV. As we keep on utilized these medications clinically, we shall mind their multifaceted mitochondrial toxicity. Mitochondrial toxicity could represent a tremendous risk of HIV-AIDS patients taking HAART treatments for a prolonged period of time. The discovery of mitochondrial toxicity could be utilized, for example, tests of mitochondrial work (e.g. ATP content, enzyme activity ), assessment of the mitochondrial membrane potential, mitochondrial RNA levels or evaluation of mitochondrial proteins, the action of cytochrome C oxidase, estimation of mitochondrial mass and investigation of mitochondrial morphology. In any case, in the interest of a clear interpretation of HAART, mitochondrial toxicity mediated insulin resistance mechanisms need to be investigated. Studies such as these may help to obviously characterize the degree of HAART mediated mitochondrial dysfunction and development of IR, and furthermore decide how focusing on mitochondria might be beneficial for the treatment of IR in HIV patients.
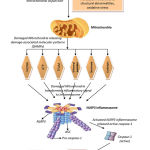 |
Figure 1: The connection between HAART medication and Mitochondrial damage, and stimulating IL-1β and IL-18-mediated inflammatory cascade. |
Figure 1. The connection between HAART medication and Mitochondrial damage, and stimulating IL-1β and IL-18-mediated inflammatory cascade.NRTIs can also bind to mitochondrial DNA polymerase gamma, which is exclusively responsible for the replication of mitochondrial DNA (mtDNA). As to NNRTI (Non-Nucleoside-Transcriptase Inhibitor) or PI (Protease Inhibitors), evidence of the damage the mitochondria .Result damaged mitochondria triggers the inflammasome NLRP3, stimulating the IL-1β and IL-18-mediated inflammatory insulin resistance cascade.
References
- WHO July 2018.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR. Recommendations and reports: Morbidity and mortality weekly report. MMWR Recomm Rep. 2015 ;64(RR-03):1.
- Muyanja D, Muzoora C, Muyingo A, Muyindike W, Siedner MJ. High prevalence of metabolic syndrome and cardiovascular disease risk among people with HIV on stable ART in southwestern Uganda. AIDS patient care and STDs. 2016;30:4-10.
- Nguyen KA, Peer N, Mills EJ, Kengne AP. A Meta-Analysis of the Metabolic Syndrome Prevalence in the Global HIV-Infected Population. PloS one. 2016;11:e0150970.
- Schulte-Hermann K, Schalk H, Haider B, Hutterer J, Gmeinhart B, Pichler K, et al. Impaired lipid profile and insulin resistance in a cohort of Austrian HIV patients. Journal of infection and chemotherapy : J Infect Chemother Title. 2016; 22:248–53.
- Mondy K, Overton ET, Grubb J, Tong S, Seyfried W, Powderly W, et al. Metabolic syndrome in HIV-infected patients from an urban, midwestern US outpatient population. Clinical infectious diseases : Clin Infect Dis . 2007;44:726–34.
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005 ;39:359-407.
- Kovacic P. Role of oxidative metabolites of cocaine in toxicity and addiction: oxidative stress and electron transfer. Med Hypotheses. 2005 ;64:350-6.
- Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008 Dec 1;1147(1):37-52.
- Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011; 14:1939-51.
- Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012 ;441:523-40.
- Sagar A, Mohanty AP, Bahal A. Zidovudine-induced myopathy: A study in Indian patients. J Neurosci Rural Pract. 201 ;1:63.
- Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med . 1990;322(16):1098-105.
- McComsey GA, O’Riordan M, Choi J, Libutti D, Rowe D, Storer N, et al. Mitochondrial function, inflammation, fat and bone in HIV lipoatrophy: randomized study of uridine supplementation or switch to tenofovir. Antiviral therapy. 2012;17(2):347.
- Sarnat HB, Marín-García J. Pathology of mitochondrial encephalomyopathies. Canadian journal of neurological sciences. 2005 May;32(2):152-66.
- Walker UA, Bickel M, Lütke SV, Ketelsen UP, Schöfer H, Setzer B,et al. Evidence of nucleoside analogue reverse transcriptase inhibitor–associated genetic and structural defects of mitochondria in adipose tissue of HIV-infected patients. Journal of acquired immune deficiency syndromes (1999). 2002 Feb;29(2):117-21.
- Gardner K, Hall PA, Chinnery PF, Payne BA. HIV treatment and associated mitochondrial pathology: review of 25 years of in vitro, animal, and human studies. Toxicol Pathol. 2014 ;42:811-22.
- Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nat Med. 1995;1:417.
- Shiramizu B, Shikuma KM, Kamemoto L, Gerschenson M, Erdem G, Pinti M, et al . Placenta and cord blood mitochondrial DNA toxicity in HIV-infected women receiving nucleoside reverse transcriptase inhibitors during pregnancy. Acquir Immune Defic Syndr. 2003 ;32:370-4.
- Gerschenson M, Nguyen V, Ewings EL, Ceresa A, Shaw JA, St. Claire MC, et al . Mitochondrial toxicity in fetal Erythrocebus patas monkeys exposed transplacentally to zidovudine plus lamivudine. AIDS Res Hum Retroviruses. 2004; 20: 91–100.
- Divi RL, Walker VE, Wade NA, Nagashima K, Seilkop SK, Adams ME,et al. Mitochondrial damage and DNA depletion in cord blood and umbilical cord from infants exposed in utero to Combivir. AIDS. 2004; 18: 1013–21.
- Divi RL, Leonard SL, Kuo MM, Walker BL, Orozco CC, Claire MC,et al. Cardiac mitochondrial compromise in 1-yr-old Erythrocebus patas monkeys perinatally-exposed to nucleoside reverse transcriptase inhibitors. Cardiovasc Toxicol. 2005; 5: 333–46.
- Maagaard A, Kyale D. Mitochondrial toxicity in HIV‐infected patients both off and on antiretroviral treatment: a continuum or distinct underlying mechanisms? J Antimicrob Chemother. 2009; 64: 901–9.
- Apostolova N, Blas-García A, Esplugues JV. Mitochondrial interference by anti-HIV drugs: mechanisms beyond Pol-γ inhibition. Trends Pharmacol Sci. 2011;32:715-25.
- Galluzzi L, Pinti M, Troiano L, Prada N, Nasi M, Ferraresi R,et al. Short communication Changes in mitochondrial RNA production in cells treated with nucleoside analogues. Antiviral therapy. 2005;10:191-5.
- Eoin R, Patrick WG. Impact of mitochondrial toxicity of HIV-1 antiretroviral drugs on lipodystrophy and metabolic dysregulation. Curr Pharm Des. 2010 ;16:3339-51.
- Jiang B, Khandelwal AR, Rogers LK, Hebert VY, Kleinedler JJ, Zavecz JH,et al. Antiretrovirals induce endothelial dysfunction via an oxidant-dependent pathway and promote neointimal hyperplasia. Toxicol Sci. 2010;117:524-36.
- Lund KC, Wallace KB. Adenosine 3′, 5′-cyclic monophosphate (cAMP)-dependent phosphoregulation of mitochondrial complex I is inhibited by nucleoside reverse transcriptase inhibitors. Toxicol Appl Pharmaco. 2008 ;226:94-106.
- Deng W, Baki L, Yin J, Zhou H, Baumgarten CM. HIV protease inhibitors elicit volume-sensitive Cl− current in cardiac myocytes via mitochondrial ROS. J Mol Cell Cardiol. 2010 ;49:746-52.
- Blas‐García A, Apostolova N, Ballesteros D, Monleón D, Morales JM, Rocha M,et al. Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology. 2010 ;52(1):115-25.
- Hernandez S, Moren C, Lopez M, Coll O, Cardellach F, Gratacos E, et al Perinatal outcomes, mitochondrial toxicity and apoptosis in HIV-treated pregnant women and in-utero-exposed newborn. AIDA. 2012;26 :419-28.
- Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384-7.
- Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev. 2010 ;31(3):364-95.
- Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2012 Feb;8(2):92.
- Martin SD, McGee SL. The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes. Biochim Biophys Acta Gen Subj. 2014 Apr 1;1840(4):1303-12.
- Shikuma CM, Day LJ, Gerschenson M. Insulin resistance in the HIV-infected population: the potential role of mitochondrial dysfunction. Curr Drug Targets Infect Disord 2005; 5:255–262.
- Barlow-Mosha L, Eckard AR, McComsey GA, Musoke PM. Metabolic complications and treatment of perinatally HIV-infected children and adolescents. J Int AIDS Soc 2013; 16:18600.
- Dagan T, Sable C, Bray J, Gerschenson M. Mitochondrial dysfunction and antiretroviral nucleoside analog toxicities: what is the evidence? Mitochondrion 2002; 1:397–412.
- Gerschenson M, Brinkman K. Mitochondrial dysfunction in AIDS and its treatment. Mitochondrion 2004; 4:763–777.
- Takemoto JK, Miller TL, Wang J, Jacobson DL, Geffner ME, Van Dyke RB, Gerschenson M. Insulin resistance in HIV-infected youth is associated with decreased mitochondrial respiration. AIDS (London, England). 2017 ;31:15.
- Araujo S, Banon S, Machuca I, Moreno A, Perez-Elias MJ, Casado JL. Prevalence of insulin resistance and risk of diabetes mellitus in HIV-infected patients receiving current antiretroviral drugs. Eur J Endocrinol. 2014; 171:545–54.
- Ben-Romano R, Rudich A, Török D, Vanounou S, Riesenberg K, Schlaeffer F, et al. Agent and cell-type specificity in the induction of insulin resistance by HIV protease inhibitors. AIDS. 2003;17:23–32
- Mondy KE, de las Fuentes L, Waggoner A, Onen NF, Bopp CS, Lassa-Claxton S, et al. Insulin resistance predicts endothelial dysfunction and cardiovascular risk in HIV-infected persons on long-term highly active antiretroviral therapy. AIDS. 2008;22:849–56.
- Butt AA, McGinnis K, Rodriguez-Barradas MC, Crystal S, Simberkoff M, Goetz MB, et al. HIV-infection and the risk of diabetes mellitus. AIDS. 2009;23:1227–34.
- Serraino D, Bruzzone S, Zucchetto A, Suligoi B, De Paoli A, Pennazza S, et al. Elevated risks of death for diabetes mellitus and cardiovascular diseases in Italian AIDS cases. AIDS Res Ther. 2010;7:11.
- Vigouroux C, Gharakhanian S, Salhi Y, Nguyen TH, Chevenne D, Capeau J, et al. Diabetes, insulin resistance and dyslipidaemia in lipodystrophic HIV-infected patients on highly active antiretroviral therapy (HAART) Diabetes Metab. 1999;25:225–32.
- Tien PC, Schneider MF, Cole SR, Levine AM, Cohen M, DeHovitz J, et al. Antiretroviral therapy exposure and insulin resistance in the Women’s Interagency HIV study. J Acquir Immune Defic Syndr. 2008;49:369–76.
- Koster JC, Remedi MS, Qiu H, Nichols CG, Hruz PW. HIV protease inhibitors acutely impair glucose-stimulated insulin release. Diabetes. 2003; 52:1695–700.
- Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707-35.
- Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617-29.
- Arnoult D, Soares F, Tattoli I, and Girardin SE. Mitochondria in innate immunity. EMBO Rep 12: 901–910, 2011.
- Schapira AH. Mitochondrial disease. Lancet 368: 70–82, 2006
- Schumacker PT, Gillespie MN, Nakahira K, Choi AM, Crouser ED, Piantadosi CA, and Bhattacharya J. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol306: L962–L974, 2014
- Taylor RW. and Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet6: 389–402, 2005
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC,et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011
- Zhou R, Yazdi AS, Menu P, and Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature469: 221–225, 2011
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al . Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36: 401–414.
- Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. 2014 :jbc-R114.
- Kim S, Joe Y, Jeong SO, Zheng M, Back SH, Park SW,et al. Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of the NF-κB and inflammasome pathways. Innate Immun. 2014; 20:799-815.
- Benetti E, Chiazza F, Patel NS, Collino M. The NLRP3 Inflammasome as a novel player of the intercellular crosstalk in metabolic disorders. Mediators Inflamm. 2013;2013.
- Fantuzzi G, Dinarello CA. Interleukin-18 and interleukin-1β: two cytokine substrates for ICE (caspase-1). Clin Immunol. 1999 ;19:1-1.
- Stienstra R, Joosten LA, Koenen T, Van Tits B, Van Diepen JA, Van Den Berg SA, et al . The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010 ;12:593-605.
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al . The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011 ;17:179.
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT,et al . Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol . 2011 ;12 :408.
- Böni-Schnetzler M, Donath MY. Increased IL-1β activation, the culprit not only for defective insulin secretion but also for insulin resistance? Cell Res. 2011;21:995-7.
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA,et al. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002; 110:851-60.
- Donath MY, Størling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T. Cytokines and β-cell biology: from concept to clinical translation. Endocr Rev. 2007 ;29:334-50.
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011 ;11:98.
- Böni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J,et al. Increased interleukin (IL)-1β messenger ribonucleic acid expression in β-cells of individuals with type 2 diabetes and regulation of IL-1β in human islets by glucose and autostimulation. J Clin Endocrinol Metab . 2008 ;93:4065-74.
- Ehses JA, Lacraz G, Giroix MH, Schmidlin F, Coulaud J, Kassis N,et al. IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc Natl Acad Sci. 2009 ;106:13998-4003.

