
Manuscript accepted on :14-Nov-2018
Published online on: 24-11-2018
Plagiarism Check: Yes
Reviewed by: Dr. Ratnadeep Ganguly
Second Review by: Archana
Final Approval by: Prof. Juei-Tang Cheng
Ni Wayan Winarti1 , Rosalina Susantio1 and Ni Putu Yuliawati2
, Rosalina Susantio1 and Ni Putu Yuliawati2
1Department of Anatomical Pathology and Faculty of Medicine, Udayana University / Sanglah Hospital Denpasar, Bali, Indonesia.
2Department of Ophthalmology, Faculty of Medicine, Udayana University / Sanglah Hospital Denpasar, Bali, Indonesia.
Corresponding Author E-mail: niwayanwinarti73@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/1590
Abstract
Retinoblastoma, a rare eye malignant tumor of childhood, is generally responsive to chemotherapy or radiation therapy, but the survivors have high risk to development of second primary tumors (SPTs) due to genetic susceptibility and/or prior radiation therapy. The SPTs predominantly occured among heritable form of Retinoblastoma and show worse prognosis than Retinoblastoma itself. A 13 years old girl underwent enucleation to remove tumor mass within her left orbit. This tumor initially appeared 4 years after the patient undergoing right bulbar enucleation and receiving chemotherapy due to Retinoblastoma in her right eye. Clinical and radiologic examination suspected the left one as a late presentation of bilateral Retinoblastoma, but microscopic examination revealed a combined Extraskeletal Chondrosarcoma. The histologic type of this SPT is rare, so does it occurance at younger age and in a survivor with unilateral Retinoblastoma wihout a history of radiation therapy. The treatment of choice is combine surgery and chemo/radiation therapy, and the prognosis is poor.
Keywords
Combined Extraskeletal Chondrosarcoma; Retinoblastoma Survivors; Second Primary Tumors
Download this article as:| Copy the following to cite this article: Winarti N. W, Susantio R, Yuliawati N. P. Combined Extraskeletal Chondrosarcoma of Left Orbit : A Rare Second Primary Tumor of Retinoblastoma Survivor A Case Report. Biomed Pharmacol J 2018;11(4). |
| Copy the following to cite this URL: Winarti N. W, Susantio R, Yuliawati N. P. Combined Extraskeletal Chondrosarcoma of Left Orbit : A Rare Second Primary Tumor of Retinoblastoma Survivor A Case Report. Biomed Pharmacol J 2018;11(4). Available from: http://biomedpharmajournal.org/?p=24218 |
Introduction
Retinoblastoma, the most common tumor of the eye in childhood, is rare and may had good prognosis. However, retinoblastoma survivors have increased risk to development of second primary tumor (SPT), whether in the Retinoblastoma eye itself or outside it. The SPTs frequently occured among hereditary form of Retinoblastoma and in patients who received prior radiation therapy. SPTs showed higher mortality rate than retinoblatoma itself. The common types of SPTs were osteosarcoma and soft tissue sarcoma, especially leiomyosarcoma.1,2 A case of SPT is reported here due to its rare histologic type and its occurance in contralateral eye of a younger age patient with unilateral retinoblastoma who never received radiation therapy.
CASE
A 13 years old girl suffered from tumor of left orbit. The tumor was initially occured as a small white plaque in her left eye when she was 7 years old. It was slowly growing tumor and it took 6 years to become a large tumor that protrude from the orbit (Figure 1). Her parents took her to Sanglah Hospital after trying to cure her with non medical alternative therapy. They did not seek hospital early because they tried to avoid same surgery like the patient previously had when she was 3 years old due to Retinoblastoma of right eye. She survives from Retinoblastoma after having right bulbar enucleation and chemotherapy, without radiation therapy. As additional information, no history of Retinoblastoma or other tumor in her family.
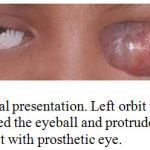 |
Figure 1: Clinical presentation. Left orbit with tumor mass that covered the eyeball and protrude from the orbit. Right orbit with prosthetic eye.
|
CT scan revealed a 3.4×4.2×4.3 cm size tumor, ful filled the orbit, with proptosis and infiltration into optic nerve and retrobulbar fat tissue (Figure 2). Radiologically, it was suspected as Retinoblastoma of left eye, a late presentation of contralateral Retinoblastoma in bilateral Retinoblastoma.
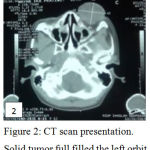 |
Figure 2: CT scan presentation. Solid tumor full filled the left orbit.
|
Left bulbar enucleation was done and the specimen was sent to pathology depertment. The tissue was 4.5×3.5×3.5 cm in size, contained tumor mass with ill-defined border, covered and infiltrated the eye ball. It was solid friable in consistency and greyish white in color (Figure 3). Microscopically, the tumor was composed by mixture of two morphological features: some area consist of well diferentiated hyaline cartilage tissue with small round undiferentiated cells, intermingled with areas of eosinophillic cells arranged in solid, strand and pseudoacinar patterns, set in myxoid matrix (Figure 4). Some of the tumor cells show immunoreactivity to vimentin and the hyaline cartilage cells is diffusely positive for S-100 protein. This morphology is consistent with combination of two types of sarcoma: Extraskeletal Mesenchymal Chondrosarcoma (EMEC) and Extraskeletal Myxoid Chondrosarcoma (EMYC).
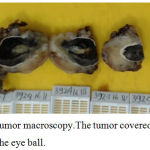 |
Figure 3: Tumor macroscopy.The tumor covered and infiltrated the eye ball.
|
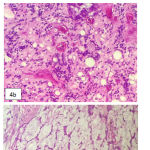 |
Figure 4: Tumor microscopy. a. Area with well differentiated hyaline cartilage tissue, b. Area with small undifferentiated cells, c. Area with eosinophillic cells set in mucoid stroma.
|
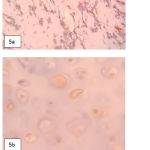 |
Figure 5: Vimentin and S100 protein immunostaining. a. Vimentine positive in eosinophillic tumor cells, b. S100 positive in hyaline certilage cells.
|
After bulbar enucleation, the patient received chemotherapy. As mention at literatures, EMEC is a grade 3 sarcoma with aggresive behaviour, hence the prognosis is poor.
Discussion
Retinoblastoma, the most common eye cancer in childhood, represents 3% of all malignancies in children under the age of 15 years.3 Retinoblastoma is caused by a mutation in the retinoblastoma gene (RB1) located on the long arm of chromosome 13 at locus 14 (13q14). This gene encodes p105 retinoblastoma protein that involves in regulating the cell cycle. Both copies of RB1 must be mutated for retinoblastoma to develop. Bilateral retinoblastoma is almost always due to a germline mutation in RB1, and 15% of patients with unilateral retinoblastoma are carriers of a germline RB1 mutation. About 6% of retinoblastoma patients have a family history of retinoblastoma.4 This reported case had history of retinoblastoma of right eye, underwent right bulbar enucleation and received chemoterapy when she was 3 years old. Detection of germline RB1 mutation was not done. Since the tumor is unilateral and no family history of retinoblastoma was noted, it is most likely a non heritable case. However, the possibility of germline RB1 mutation can not be excluded.
In recent years, there have been dramatic changes in the management of intra orbital retinoblastoma. The treatment paradigm changed from systemic to local therapy, in order to preserve vision and prevent tretment induced SPT. When the disease has extended outside the eye, the chances of cure are significantly lower. Some predictive factors must be evaluated pathologically, such as, invasion of the choroid, the optic nerve, and the sclera. These information is used to decide the need of adjuvant therapy. Patient with overt extraocular lesion are treated with higher dose neoadjuvant therapy followed by delayed enucleation and adjuvant therapy.5 This malignancy leads to metastatic disease and death in 70% of children in lower and middle economic countries6, but in developed countries the cure rate is about 95-98%.3 This reported patient underwent right bulbar enucleation, continued with six series of systemic chemotherapy, without radiation therapy. Pathological review to evaluate pathologic predictive factors can not be done, because in our pathology laboratory, histologic slides are only archived for 10 years. Except loss of visual function of the right eye, no reccurence nor related complication in the right eye was noted until present days.
Retinoblastoma survivors are at high risk to have SPTs. The cumulative incidence for SPTs 50 years after the diagnosis of Rb is 36% (95% CI 31-41%).7 This SPTs mainly occured due to RB1 gene mutasion. RB1 gene, actually not only has a role in cell cycle regulation, but also in cellular differentiation, regulation of apoptosis and preservation of chromosomal stability. Loss of RB1 function contribute to cancer formation, either in initiation or progression stage.4,8 The risk of SPTs development is higher in heritable than non heritable cases. MacCarthy, et al. (2013) reported that the Standardised Incidence Ratios (SIRs) of all types of SPTs were 13.7 (CI 95% 11.3-16.5) in heritable cases. It was 8-10 times than non-heritable cases.2 This reported case is one of SPT case that occured in a possibly non heritable Retinoblastoma.
MacCharty, et al. (2013) also analyzed SPTs incidence based on the affected region: head and neck region (radiation field) and outside head and neck region. The SIRs were 2-3 times higher in head and neck region than outside head and neck region. Further more, the SIR for head and neck region tumors for heritable cases is about 25 times that for those outside this region in the non-heritable cases. This analysis suggests that there is a possible multiplicative effect of having an RB1 gene mutation and being irradiated.2 In this reported case, the SPT occured in head and neck region in a patient that never received radiation therapy.
Three most frequent SPTs in retinoblastoma were osteosarcoma, leiomyosarcoma and skin melanoma. Other rare types include other sarcoma, carcinoma and brain tumors. Median age at diagnosis of SPTs was 13 years. The median age at which SPTs occurred inside the radiation field was younger than that for SPTs occurring outside the radiation field or in patients who did not undergo irradiation.9 The SPT in this reported case was initially occured when the patient was 7 years old. It appeared at younger age than in most cases who did not received radiation therapy.
Sarcomas occurred more commonly inside the radiation field. Melanomas, lipomas, leukemias, and lymphomas occurred more commonly outside the radiation field or in patients who did not undergo irradiation.9 This reported case is a sarcoma. It is infrequently appeared in pasients who did not undergo irradiation.
Pathologically, histological type of this SPT case is also infrequent because microscopic examination showed combination of two different types of sarcoma in one tumor. Some microscopic areas show features that fit to diagnosis EMEC and some other areas fit to diagnosis EMYC.
EMEC is a rare variant of chondrosarcoma. Although may occur in young children, the peak incidence was in 15-35 years of age, and more frequent in female than male. Only about 10% of EMEC was located in orbit, and it tend to produce exophthalmus, vision blurring and orbital pain. Microscopically, EMEC is composed by sheets of undifferentiated cells with an abrupt transition with small well-define nodules of well-differentiated hyaline cartilage. The undifferentiated cells show ovoid or elongated hyperchromatic nuclei and scanty, poorly outlined cytoplasm. S100 immunostainning show difuse positivity in cartilage areas and isolated cell positivity in undifferentiated cells.10 Pathological features of this reported case fit the literature (Figure 4 and 5).
Unsimilar with EMEC, though named as chondrosarcoma, EMYC actually is not variant of chondrosarcoma. WHO categorized it as a tumor of uncertain differentiation because there is a paucity of convincing evidence of cartilaginous differentiation. EMEC occurs most frequently in the deep tissues of extremities, in patient older than 35 years old, and more frequent in male. Some cases with EMEC have been reported in unusual location, including the mediastinum, vulva, central nervous system and heart.10 EMYC in this case occured at unusual location, in a young female patient.
Microscopically, EMYC shows multinodular pattern, composed by cells that round or slightly elongated in shaped, with uniform shape and size, separated by mucoid material. The cells have small hyperchromatic nuclei and deeply eosinophillic cytoplasm reminiscent of chondroblasts, and arranged in short anastomosing cords, strands or pseudoacini, often creating a lace like appearance. The mucoid material consists largely of chondroitin 4 sulfate, chondroitin 6-sulfate and keratin sulfat, that can be visualized better with colloidal iron stain and the alcian blue stain. Immunohistochemical findings are not consistently positive, excep for vimentin.10 The morphology of this reported case is fit to the literature. The tumor cells are positive for vimentine, but we did not do histochemical staining to highlight the mucoid material.
Cytogenetically, EMEC showed evidence of recurrent HEY1-NCOA2 [(8;8)(q21;q13)] fusion, whereas most EMYC showed translocations t(9;22)(q22;q12) and t(9;17)(q22;q11).10 Since we did not do cytogenetic and molecular tests, we do not know whether this case arise from combination of those two types of alteration, or does it result from different alteration.
The treatment of choice of this sarcoma is combined radical surgery and chemotherapy or radiotherapy. EMEC is a grade 3 malignant tumor, with aggressive clinical course and highly metastasized, particularly to the lung. The 5- and 10-YSR are 54.6% and 27.3% respectively. Whereas, EMYC is a grade 1-2 malignant tumor, in which increasing patient age, large tumor size and proximal tumor loacation as predictive value for adverse outcome. Although slowly growing, EMYC also has propensity for pulmonary metastasis.10 This case was concluded as a grade 3 tumor because of its EMEC component. This patient received chemotherapy after left bulbar enucleation, but the prognosis is poor.
Conclusion
A case of Combined Extraskeletal Chondrosarcoma of left orbit has been reported here. It is a very rare type of SPT, occured at younger age and in a survivor of unilateral Retinoblastoma wihout a history of radiation. After bulbar enucleation, the patient received chemotherapy. It is an aggressive malignant tumor with poor prognosis.
Since Retinoblastoma survivors have high risk to development of SPTs, and the SPTs generally showed higher mortality than Retinoblastoma itself, hence long term follow up of Retinoblastoma survivors is highly recommended.
Acknowledgment
This case had been previously presented as a poster presentation at Annual Pathology Meeting of Indonesian Association of Pathologist in Bali, Indonesia, in 2017, but the full paper never been published in other journal.
Conflict of Interest
The authors declare no conflict of interest or any financial support, and had got permission from patient parents to publish this case.
References
- Kleinerman R. A., Yu C. L., Little M. P., Li Y., Abramson D., Seddon J and Tucker M. A. Variation of Second Cancer Risk by Family History of Retinoblastoma Among Long-Term Survivors. Journal of Clinical Oncology. 2012;30(9):950-7.
CrossRef - MacCarthy A., Bayne A. M., Brownbill P. A., Bunch K. J., Diggens N. L., Draper G. J., Hawkins M. M., Jenkinson H. C., Kingston J. E., Stiller C. A. C. A., Vincent T. J and Murphy M. F. G. Second and Subsequent Tumours among 1927 Retinoblastoma Patients Diagnosed in Britain 1951–2004. British Journal of Cancer. 2013;108:2455-63.
CrossRef - Yun J., Li Y., Xu C. T and Pan B. R. Epidemiology and RB1 Gene of Retinoblastoma. J. Ophthalmol. 2011;4(1):103-9.
- Chen J. J and Allen R. C. Secondary Tumors After Hereditary Retinoblastoma: A Case of Orbital Leiomyosarcoma 50 Years After Initial Enucleation and Radiation Therapy. org. November 28, Available from: http://EyeRounds.org/cases/142–‐leiomyosarcoma.htm. 2011.
- Chantada G and Schaiquevich P. Management of Retinoblastoma in Children: Current Status. Pediatric Drugs. 2015;17:185-98.
CrossRef - Dimaras H., Kimani K., Dimba E. A. O., Gronsdahl P., White A., Chan H. S. L and Gallie B. L. Lancet. 2012;379:1436-46.
CrossRef - Lohmann D. Retinoblastoma. In: Ahmad S.I. (Editor). Diseases of DNA Repair. Landes Bioscience and Springer Science+Business Media. 2010;220-1.
CrossRef - Burrkhart D. L and Sage J. Cellular Mechanisms of Tumour Suppression by The Retinoblastoma Gene. Nat Rev Cancer. 2008;8(9):671-82.
CrossRef - Woo K. I and Harbour W. Review of 676 Second Primary Tumors in Patients with Retinoblastoma. Association Between Age at Onset and Tumor Type. Arch Ophthalmol. 2010;128(7):865-870.
CrossRef - Goldblum J. R., Folpe A. R and Weiss S. W. (editors). Enzinger & Weiss’s Soft Tissue Tumor, sixth edition. Philadelphia:Elsevier Saunders. 2014;922-6 and 1045-52.

