
Manuscript accepted on :28-Aug-2018
Published online on: 04-09-2018
Plagiarism Check: Yes
Reviewed by: Peter Uetz
Second Review by: Dr. Amit Jain
Final Approval by: Prof. Juei-Tang Cheng
Rohini Mugur, Smitha P. S and Pallavi M. S.
Department of Computer Science Amrita School of Arts and Sciences Amrita Vishwa Vidyapeetham Mysuru, Karnataka, India.
Corresponding Author E-mail: surendararavindhan@ieee.org
DOI : https://dx.doi.org/10.13005/bpj/1540
Abstract
The Protein complexes from PPIs are responsible for the important biological processes about the cell and learning the functionality under these biological process need uncovering and learning complexes and related interacting proteins. One way for studying and dealing with this PPI involves Markov Clustering (MCL) algorithm and has successfully produced result, due to its efficiency and accuracy. The Markov clustering produced result contains clusters which are noisy, these wont represent any complexes that are known or will contains additional noisy proteins which will impact on the correctness of correctly predicted complexes. And correctly predicted correctness of these clusters works well with matched and complexes that are known are quite less. Increasing in the clusters will eventually improve the correctness required to understand and organize of these complexes. The consistency of experimental proof varies largely techniques for assessing quality that have been prepared and used to find the most suitable subset of the interacting proteins. The physical interactions between the proteins are complimented by the, amplitude of data regarding the various types of functional associations among proteins, which includes interactions between the gene, shared evolutionary history and about co-expression. This technique involves the facts and figures from interactions between the proteins, microarray gene-expression profiles, protein complexes, and practical observations for proteins that are known. Clusters communicate not only to protein complex but they also interact with other set proteins by this, graph theoretic clustering method will drop the dynamic interaction by producing false positive rates.
Keywords
Cluster MCL; Gene; Interaction; MCODE; Network; Protein; RRW Algorithms
Download this article as:| Copy the following to cite this article: Mugur R, Smitha P. S, Pallavi M. S. Predicting the Functions of Unknown Protein by Analyzing Known Protein Interaction: A survey. Biomed Pharmacol J 2018;11(3). |
| Copy the following to cite this URL: Mugur R, Smitha P. S, Pallavi M. S. Predicting the Functions of Unknown Protein by Analyzing Known Protein Interaction: A survey. Biomed Pharmacol J 2018;11(3). Available from: http://biomedpharmajournal.org/?p=22186 |
Introduction
T Stoddard, B.L et.al1 Protein composites are answerable for most of vital biotic procedures within the cell. It is also important in considering the technics under these biological programs requires detecting for analyzing protein complex and their related proteins.
Dartel, P L, et.al2 Systematic experiments of functional genomics is considered for screening interesting genes. Protein–protein interactions experiments are more interesting because interacting proteins well collaborate on a common purpose.
Madden, T L et.al3 Similarly functioned genes are probably expressed together. By analyzing cluster of gene expression are used for predicting function of unknown proteins .
Hishigaki, H et.al4 The primary function of protein is protein-protein interaction which is necessary to understand. As the size of protein interaction keeps on increasing, here the interaction between proteins takes place as a cluster and effectiveness in finding significant complex is performed.
Smith, G, R et.al5 This paper work has shown that the information of 3D structures can be utilized to anticipate the Protein interaction with an efficient way and they cover that they are superior predictions on non-architectural proof and are taken as primary entity.
Canutescu, A et.al6 The paper has proposed about the past advancement that are made in predicting the structure of clusters that are joined when the alike components are known .
Letovsky, S et.al7 After the completion of sequencing a number of genomes, now it’s focused on proteomics. An advanced proteomics technologies like two-hybrid test are leading in to the huge data sets of interaction between the protein which can be designed as a networks of it , and the major issue is to discover protein combinations within network.
Stanley Letovsky et.al8 Expression profiling in interaction of protein are some of the high throughput functional genomics techniques which produce new datasets which provide more options for interpretation of function.
Stanley Letovsky et.al9 Markov Random Field (MRF) formalism are used to provide a more robust probabilistic solution. This technique used for image analysis i .e, for image restoration and segmentation. Here we can use for segmenting protein-interaction network into sub graphs that share to similar label
Avazquez, A et.al10 In the, let F be the total number of functions in functional classification scheme. In principle, to each protein should assigned some of the functional classes drawn from these classes. So the subset of the proteins in the network from the functional can be used.
Minghua Deng, et.al11 Here a mathematical method is used for interactions of protein, Bayesian analysis is used for assigning functions to proteins. Posterior probabilities for unannotated protein is estimated by Gibbs sampler.
M Deng, et.al12 Maximum likelihood estimation (MLE) methods are used for determining the reliability of several protein interaction data sets .
Rentaro Saito, et.al13 The availability of each candidate interaction between the proteins plays an important role. “Interaction generality” measure (IG1) which can be used for assigning the reliability .
Brun, C, et.al14 The direct interaction between protein partners are likely to distribute same kind of functions with it. It has shown that 70–85% of proteins will have minimum one function with its interacting protein partner.
Alm E et.al15 The non-homology based approaches for the purposes of efficient annotation provides an alternative high throughput. These ways and means are built by relationship, in which the proteins are practically connected by either trial conducted or by estimating means.
Comeau, S,R et.al16 Anticipating the interaction between the proteins is one of the toughest and accost problems in identifying function of genomics as its helps in diagnosing the functional defect in the one’s body.
Arnau, V et.al17 The primary function of protein is PPI which is necessary to understand cellular function. The experimental on PPIs have resulted in a huge number of interactions between proteins that yields to anticipate the protein complexes from PPI network. High throughput experiments will produces are repeatedly combined with both correct and incorrect values so by this prediction protein complexes becomes harder.
Janscn, R et.al18 The bacteria called yeast interatomic can hold up to eighty thousand protein network Interactions. This estimation is based on the integration of data sets obtained by various methods like mass genetic studies.
Minghua Deng et.al19 Fraction of proteins that have the related functions are considered. They
Trupti Joshi, et.al20 The Experiment on Saccharomyces cerevisiae by combining biological data on micro array gene expression for finding functional description of hypothetical proteins can be done using statistical model.
provide an equal weight to intra-function interactions between the classes of proteins.
Yu chen dong, et.al21 For the Gene Ontology (GO) biological processes which have Reliability scores the proteins with the unknown functions can be assigned. This is better than MIPS which have less details
Lukasz Salwinski, et.al22 The dependence on the practically examined on a genome roused development of data quality for assigning methods. Database provided development to the database schema which permits to catch more detailed information on the molecular interactions
Elena Nabieva et.al23 Analyzing protein interaction maps should be the basics for the further organization of the cell and provide support in such a way that do not hide protein functions.
Michele Leone, et.al24 The data that are available in a graph-like format in the online structure, with graph sites with links represents the interaction between two proteins with protein names.
Hon Nian, Chua et.al25 Consequences of ”indirect functional association” in existing interaction in protein network data in the “Saccharomyces genome” is taken and new technic which account indirect functional association for prediction of protein function is considered.
Mintseris et.al26 The study of physics in Biology of interactions between protein and docking will have effects on most of complex cellular signaling processes.
Sharan, R et.al27 The functional definition of proteins was primary issue in post‐genomic era. The recent interaction between the protein in network data of many model types has arouse the growth of computational methods for inferring related data to clarify protein function .
Chua,H.N et.al28 Understanding the Protein complexes are principal function to implement & to understand the ideologies of organizations of cells as it includes masses of interaction between protein (PPI) networks.
Jungsh et.al29 A complex of protein is a proteins cluster that interact with each other at the same instance, the results of interaction between protein says that data helped us to improve computational ways for complex protein predictions.
Shrihari, S et.al30 The Protein campuses are chief body to develop many biological methods in cell and complete body, like signal conversion, gene expressions, and molecular transmissions.
Ozawa, Y et.al31 The forth put of this paper says that the rate of production for detecting the protein-protein interactions resulted in vast relationship of networks, and permitted to computationally find the families of proteins.
Habibi.M et.al32 The Protein clusters play a vital role in cellular mechanisms and in recent years several ideas and methodology have been proposed and presented and made available to predict complexes of protein in a network of protein.
Li, Z et.al33 In post genomic era detecting Protein – Protein Interaction was a challenging task. As a result of accumulating amount of protein interaction data are feasible and protein complexes can also be identified from PPI networks, In recent studies detecting protein complexes are purely on the observation of that heavy region of PPI networks which is correlated to protein complexes.
Xie, Z., kwoh et.al34 Group of proteins are the responsible in solving the secrets of group of cells and also find function in human body. The system AP-MS show higher rate throughput screening in gauging the direct and indirect proteins which are connected, and the results includes both positives and negatives.
Maruyama, O et.al35 The cluster of physical interaction proteins aggregate the basic functional units are responsible for performing biological processes involved in cells. A renovation of the entire set of compound is cardinal to apprehend the organization functions within the cells.
Zhang, O, C et.al36 The practicable broadcasting of readable open frames are encoded in the genome is the main function in yeast genomics. When the adjacent interacting protein is known then identifying the functions of proteins will be easier.
Srihari.S et.al37 The survey conducted has reviewed, classified that the computational methods to evolve for identifying of protein complex from Protein interaction networks.
Devasia,et.al38 has proposed a Prediction Method, for calculating performance of Students. The Prediction approachs gives a good results
Tatsuke, D et.al39 In this paper work, based on assembled conception which is on account of Protein interaction networks, Protein interaction data and Gene Ontology resource. After building ontology which are accredited networks, it is suggested that a unique approach called CSO (clusters based on web structure and ontology) works well which leads to produce an accurate result.
Zhang, Y et.al40 The proposed paper explains a methods of allowing the function of analyzing the graph of adjacency in a PPI network. This tells graph neighbors share function with direct nodes than indirect nodes.
Shrihari, S et.al41 In this paper the author has provide with SCWRL programs alike that the method was broadly used because of its high rate, efficiency, and its simplicity. This presented that, the problems that are found in the side-chain prediction is referred from the results of graph theory. The method used here show that the side chains are characterized as vertices in undirected graph.
Suresh et.al42 has proposed a prediction method for student Academic dashboard. The predictive models are effective and useful.
Algorithm
MCL-CA
When the protein performance is matched ,the Protein Docking Benchmark , shows improved result with the decreased with comprehensive recording functions ,as a result the new data shows its positive result on antibiotic-antigen complexes, also most clustering predictions by determining the regions of antibodies without manual intermediation.
FS Weight
By using (FS Weight), all adjacent and unconnected Protein interactions are weighted which evaluates the performance of functional association for the further processing the interaction with lower weight is removed from the network. An another approach can be a novel algorithm that works In the changed network searching for the cliques, and unify those obtained cliques to form clusters using a “partial clique merging” technique. The experiments showing that when the algorithm is applied in finding the composite network, it performs very well on interacted and modified networks.
MCODE
MCODE is one of the widely used method for the computational purposes in identifying the complex PPI networks. The algorithm works step by step in two levels, one is being the vertex weighting and secondly prediction of complex networks, and an elective third stage for post-processing (if it is required).
The molecular complex detection algorithm (MCODE) works in three levels Once the computing of the weights is completed, the algorithm travel through these weighted graph. After computing the weights the algorithm traverses through the weighted graph in a desirous manner to detach closely linked areas.
SPIN and MEIs
For the prediction of complex network, Author used a technique that uses the” interface and structural data pairs of protein” in predicting the complex network, meanwhile, a “simultaneous protein interaction network “(SPIN) is introduced to insist on “mutually exclusive interactions”(MEIs) from the interconnecting interfaces. After the completion of SPINs, there by a protein complex is formed and to predict this complex network a naive clustering algorithm is used. The results showed that the applied method bangs the simple “PPIN-based method“in eliminating the incorrect positive proteins in the complex formation and this discounting the competition among MEIs helps in rising accuracy rate of the general computational which involves protein interaction.
Co-complexes Score
In this article, without depending on the knowledge of known complexes, we propose a novel unproven approach. Our system calculates the similarity among two proteins, and the similarity is evaluated by a co-complexes score or C2S in short. In specific, the method relay on the log-likelihood ratio of 2 proteins which is being co-complexes to be drawn unsystematically, and we then determine protein complexes by using a classified clustering algorithm.
MCL-CA
The method MCL-CA, is connected by core-attachment which is mainly used to refine the clusters obtained by MCL-CA and also gauged efficiency of method on different datasets and also matched the excellence of our regulated complex networks which is formed by MCL. The outcome show that “our approach significantly improves the accuracies of predicted complexes when matched with known complexes. The result of the MCL-CA is possible to shield a huge number of known complexes than the MCL”.
CFA
This paper proposes a technique which gives appropriate protein complex prediction, that is ,CFA is a sub graph with the connectivity number on it. we will estimate results of CFA with the help of various available protein networks, based on the two protein complex standard data sets “MIPS and Aloy”, having 1142 and 61 known compounds respectively. We then compare CFA data with some “current protein complex prediction methods (CMC, MCL, PCP and RNSC) in terms of recall and accuracy”. CFA predicts add-on complexes correctly at inexpensive level of precision.
RRW Algorithm
RRW algorithm is one of the algorithm which is mainly used to Predicting the protein complex interaction in the network, which constantly expands an existing bunch of proteins in a “stagnant vector of a random walk” with resumes the cluster where the proteins are similarly weighted. In the group, the expansion of all the proteins inside the cluster have equally influenced in determining of newly and added proteins into the cluster. For the further processing we extend the RRW algorithm by introducing an unsystematic walk with restarting a group of proteins, each one in the group weighted by the sum of the stability for the proof of the direct physical interactions involved in the protein. This resulted in the rising up of an algorithm is called” NWE (Node-Weighted Expansion of clusters of proteins)”. The interactive resource in the network is got from the WI-PHI database.
Markov Clustering
Markov clustering (MCL) “is a firm, and highly mountable graph clustering method. Grouping of protein sequences, MCL has provided an effective way of clustering large protein-protein interaction networks due to its scalability. MCL works by unsystematic walks (in flow) to extract densely regions from the protein web”. To match with the flow, “MCL repeats operates the adjacency or nearby matrix in the network using two operators, they are expansion and inflation, which controls the distribution and viscosity of the flow”.
Merging Maximal Cliques (CMC)
“Merging Maximal Cliques (CMC)” clustering – CMC works based on combination of maximal cliques mined from the interaction network. CMC includes scores for interactions and improves on earlier clique-merging approach, with “C Finder Local Clique Merging Algorithm(LCMA)” that is applied only on unscored networks.
MCL, CMC, Cluster ONE
Ensemble clustering includes multiple algorithm such as (MCL, CMC, Cluster ONE) by adopting voting on the basis of scoring. By incorporating complementary data with the analysis of PPI overcome noise in the data which will help in prediction of PPI complex structure.
Functional Similarity Weight
D( u ,v)=∣Nu Δ N v∣∣ Nu ∪ N v∣+∣ Nu ∩ NV ∣,
The CD-distance between two proteins u and v is given by
Functional similarity weighted averaging
“The χ2 statistics of function j for protein i is computed by
Si(j)=(ni (j)−ei(j))2ei(j)”
Majority
In majority”all the proteins which are neighbors are taken and add the number of times each annotation that arrive for every protein as explained in Schwikowskiet”.
Neighborhood
The score of an each protein in a specific function is given by the χ2-test.
The Functional flow Algorithm
Simplifies the “guilt by association” principle to protein cluster which have or do not have interaction.
Functional linkage graph- “protein-protein interaction data using a graphical method called a functional linkage graph in which an edge (link) between two nodes (proteins) represents that they might share the same function”.
Propagation probabilities-“Probability label of a protein relays on its neighbors which in turn depend on their neighbors, we would like a difficult method of increasing our estimate for labeling probability of a protein”.
MRF
Known proteins can be divided into different classes on the basis of their functionality. The interaction between known two proteins can be arranged into one of the three groups:” (1, 1), (1, 0) and (0, 0)”
Here, Z (θ) = partition function in theory of MRF.
Global optimization principle: “a score is associated to any given assignment of functions for the set of proteins which are not classified”. The score will be less in configurations which increases the presence of the same functional detection in interacting proteins.
Self-consistent test-concerns the presence of errors in the protein network topology. The protein interactions data obtained from the hybrid experiments will have a huge amount of false positives and negatives.
Tolerance Error Checking
Which checks for tolerance error that has to satisfy the score.
Indirect Functional Association
Here it is focused on checking “protein shares function” with its “level-2 neighbors” instead of its “level-1 neighbor” Functional similarity weight. Estimation of two proteins is done by functional similarity.
Czekanowski-Dice Distance (CD-Distance)
It’s as a metric for functional linkage. The (u,v) is measured as the distance between two proteins and it is given by combining reliability of experimental sources.
Bayesian Analysis
This is used to estimate probability of the protein that has functions, by the fraction of all the proteins having that function.
A-priori Probabilities
Is used for the prediction which is based on the idea of “guilt by association”. when the interaction between the proteins take place the hypothetical protein X contain known function which divide same function among themselves with the probability monitored with high throughput data with the relationship between X and associated protein .The reliability score of function F is given by,
Neighborhood Algorithm
The protein of interest is assigned the function with the highest χ2 value is assigned to protein of interest with the functions of all n-neighbor proteins.
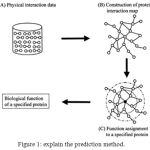 |
Figure 1: explain the prediction method.
|
The black dotted circle denotes a query protein to which function is predicted and White colored circles denotes proteins.
(20.1) Assigning functions to a query protein. This is performed depending on the functions of connected proteins on the protein map.
(20.2) Physically interacted protein data placed in the public databases.
(20.3) Construction of the protein interaction map by combining all physical interaction data.
Functional similarity weight
(21.1) “The distance between two proteins u and v is represented by D (u,v)=∣NuΔNv∣∣Nu∪Nv∣+∣Nu∩aNv∣” (21.2)Functional similarity weighted averaging The χ2 statistics for protein I and function j is composed by “ Si(j )= (ni(j)−ei(j))2ei(j)”.
Majority
All neighboring proteins are considered and they are added the number of times each similar functions that occur for each protein. In weighted interaction graphs, known methods are extended by considering a weighted sum in that place. The score for the protein of a particular function is the corresponding sum.
Gen Multicut
In ” multiway K –cut”, the primary process is to screen a graph in such a way that each of k terminal nodes bare related to various subset of the partition and so that the weighted number of edges that are ‘cut’ in the process are reduced.
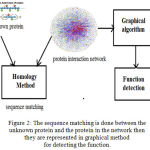 |
Figure 2: The sequence matching is done between the unknown protein and the protein in the network then they are represented in graphical method for detecting the function.
|
Conclusion
Predicting the function in post genomic era is one of the difficult task. By using homological method unknown proteins sequence are matched with known proteins in protein interaction network and then they are represented in graph for predicting functions.
References
- Stoddard B.L & Koshland D.E. Prediction of the structure of a receptor–protein complex using a binary docking method. Nature. 1992;358(6389):774-776.
CrossRef - Bartel P.L., Roecklein J.A., Gupta S.D., Fields S. A protein linkage map of Escherichia coli bacteriophage T 7. Nature Genet. 12:72.
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z.,Miller W. and Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. 1997.
- Hishigaki H., Nakai K., Ono T., Tanigami A & Takagi T. Assessment of predictive accuracy of protein function from protein–protein interaction data. Yeast. 2001;18(6):523-531.
CrossRef - Smith G.R & Sternberg M.J. Prediction of protein–protein interactions by docking methods. Current opinion in structural biology. 2002;12(1):28-35.
CrossRef - Canutescu A. A., Shelenkov A. A & Dunbrack R.L. A graph‐theory algorithm for rapid protein side‐chain prediction. Protein Science. 2003;12(9):2001-2014.
CrossRef - Letovsky S & Kasif S. Predicting protein function from protein/protein interaction data: a probabilistic approach. Bioinformatics. 2003;19(1):197-204.
CrossRef - Letovsky S, Kasif S. Bioinformatics. 2003 3 July;19(1):197–204. https://doi.org.
CrossRef - Letovsky S, Kasif S. Bioinformatics. 2003 3 July;19(1):197–204. https://doi.org/10.1093/bioinformatics/btg1026.
- Vazquez A, Flammini A, Maritan A. Vespignani(Submitted on 24 Jun 2003).
- Deng M, Zhang K, Mehta S, Fengzhu C.S. Molecular and Computational Biology Program, Department of Biological Sciences. 2003.
- Deng M., Sun F., Chen T. Pacific Symposium on Biocomputing. 2003;8:140-151.
- Saito R,Suzuki H, Hayashizaki Y. Bioinformatics,12 April .2003;19(6):756–763. https://doi.org/10.1093/bioinformatics/btg070.
CrossRef - BrunC., et al. Functional classification of proteins for the prediction of cellular function from a protein–protein interaction network. 2003.
- Alm E and Arkin A.P. Biological networks.Curr. Opin.Struct. Biol. 2003; 13:193–202. Ashburner M., Ball,C.A., Blake J.A., B.
- Comeau S.R., Gatchell D. W., Vajda S & Camacho C. J. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20(1):45-50.
CrossRef - Arnau V., Mars S & Marín I. Iterative cluster analysis of protein interaction data. Bioinformatics. 2004;21(3):364-378.
CrossRef - Jansen R & Gerstein M. Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Current opinion in microbiology. 2004;7(5):535-545.
CrossRef - Deng M., Tu Z., Sun F., Chen T. Bioinformatics. 2004;20(6):12 April 895–902. https://doi.org/10.1093/bioinformatics/btg500
CrossRef - Joshi T., Chen Y., Jeffrey M., Nickolai B. A., Xu D. Digital Biology Laboratory, Computer Science Department, University of Missouri-Columbia, Columbia, MO, USA. 2004.
- Chen Y., Xu D. Nucleic Acids Research. 2004;32(21):1 January 6414–6424.
- Salwinski L., Christopher S.M., Adam J.S., Frank K.P, James U.B., Eisenberg D. Nucleic Acids Research. 2004 January 1;32(1):449–451.
CrossRef - Nabieva E., Jim K., Agarwal A., Chazelle B., Singh M. Bioinformatics. 1 June 2005;21(1):302–310. https://doi.org/10.1093/bioinformatics/bti1054.
CrossRef - Leone M., Pagnani A. Bioinformatics. 2005;15 January 21(2):239–247. https://doi.org/10.1093/bioinformatics/bth491.
CrossRef - Nian H.C., Wing-Kin Limsoon S.W. Bioinformatics. 1 July 2006;22(13):1623–1630. https://doi.org/10.1093/bioinformatics/btl145.
CrossRef - Mintseris J., Pierce B., Wiehe K., Anderson R., Chen R & Weng Z. Integrating statistical pair potentials into protein complex prediction. Proteins. Structure. Function and Bioinformatics. 2007;69 (3):511-520.
CrossRef - Sharan R., Ulitsky I & Shamir R. Network‐based prediction of protein function. Molecular systems biology. 2007;3 (1):88.
CrossRef - Chua H. N., Ning K., Sung W. K., Leong H. W & Wong L. Using indirect protein–protein interactions for protein complex prediction. Journal of Bioinformatics and computational biology. 2008;6(03):435-466.
CrossRef - Jung S. H., Hyun B., Jang W. H., Hur H. Y & Han D. S. Protein complex prediction based on simultaneous protein interaction network. Bioinformatics. 2009;26(3):385-391.
CrossRef - Srihari S., Ning K & Leong H. Refining Markov Clustering for protein complex prediction by incorporating core-attachment structure. Genome Informatics. 2009;23(1):159-168.
CrossRef - Ozawa Y., Saito R., Fujimori S., Kashima H., Ishizaka M., Yanagawa H & Tomita M. Protein complex prediction via verifying and reconstructing the topology of domain-domain interactions. BMC Bioinformatics. 2010;11(1):350.
CrossRef - Habibi M., Eslahchi C & Wong L. Protein complex prediction based on k-connected subgraphs in protein interaction network. BMC Systems Biology. 2010;4(1):129.
CrossRef - Habibi M., Eslahchi C & Wong L. Protein complex prediction based on k-connected subgraphs in protein interaction network. BMC Systems Biology. 2010;4(1):129.
- Li X., Wu M., Kwoh C. K & Ng S. K. Computational approaches for detecting protein complexes from protein interaction networks: a survey. BMC genomics. 2010;11(1):S3.
CrossRef - Xie Z., Kwoh C. K., Li X. L & Wu M. Construction of co-complex score matrix for protein complex prediction from AP-MS data. Bioinformatics. 2011;27(13):i159-i166.
CrossRef - Maruyama O & Chihara A. NWE: Node-weighted expansion for protein complex prediction using random walk distances. Proteome science. 2011;9(1):S14.
CrossRef - Zhang Q.C., Petrey D., Deng L.L.Q., Shi Y., Thu C. A., Bisikirska B & Califano A. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490(7421):556.
CrossRef - Srihari S & Leong H. W. A survey of computational methods for protein complex prediction from protein interaction networks. Journal of Bioinformatics and computational biology. 2013;11(02):1230002.
CrossRef - Tatsuke D & Maruyama O. Sampling strategy for protein complex prediction using cluster size frequency. Gene. 2013;518(1):152-158.
CrossRef - Zhang Y., Lin H., Yang Z., Wang J., Li Y & Xu B. Protein complex prediction in large ontology attributed protein-protein interaction networks. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2013;10(3):729-741.0
- Rihari S., Young C. H., Patil A & Wong L. Methods for protein complex prediction and their contributions towards understanding the organization, function and dynamics of complexes. FEBS letters. 2015;589(19):2590-260.
- Devasia T., Vinushree T. P & Hegde V. Prediction of students performance using Educational Data Mining. In Data Mining and Advanced Computing (SAPIENCE). International Conference on .IEEE. 2016 March;91-95.
- Suresh A., Rao H. S & Hegde V. Academic Dashboard—Prediction of Institutional Student Dropout Numbers Using a Naïve Bayesian Algorithm. In Computing and Network Sustainability. 2017;73-82. Springer, Singapore.
CrossRef

