
Manuscript accepted on :26-02-2026
Published online on: 20-05-2026
Plagiarism Check: Yes
Reviewed by: Dr. Manju Jakhar
Second Review by: Dr. Priya Gayathri
Final Approval by: Dr. Patorn Piromchai
Swapnil Das1 , Fatiha Sultana Fiha1, Irfan Hoque Tanjid1, Rohit Datta1, Dip Mohajon1 and Pritesh Ranjan Dash2*
, Fatiha Sultana Fiha1, Irfan Hoque Tanjid1, Rohit Datta1, Dip Mohajon1 and Pritesh Ranjan Dash2*
1Department of Pharmacy, University of Science and Technology Chittagong, (USTC), Chittagong, Bangladesh
2Department of Pharmacy, ASA University Bangladesh, Shyamoli, Dhaka, Bangladesh
Corresponding Author: pritesh.ju@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/3427
Abstract
Acid sphingomyelinase deficiency (ASMD), also referred to as Niemann-Pick disease types A and B, is a rare lysosomal storage disorder. Mutations in the SMPD1 gene result in insufficient activity of the acid sphingomyelinase (ASM) enzyme, which causes acid sphingomyelinase deficiency (ASMD). This enzyme is necessary for the breakdown of sphingomyelin, a type of sphingolipid found in the membranes of animal cells, particularly in the outer leaflet of the plasma membrane. Without this enzyme, sphingomyelin cannot be broken down, leading to its accumulation in organs such as the liver, spleen, lungs and brain. Historically, acid sphingomyelinase deficiency (ASMD) has been classified as Niemann-Pick disease (NPD) types A and B. Type A is caused by severe deficiency in ASM leading to neurodegeneration and organ enlargement. This type typically results in death by the age of three. In contrast, type B results from partial ASM deficiency and patients typically survive but face various complications throughout their lives. Currently, treatment options for this disease are limited, although many therapeutic approaches are gradually developing. Early diagnosis and effective management can minimize the complications of this disease and increase life expectancy. So, this review aims to provide a comprehensive understanding of ASMD, including its epidemiology, pathogenesis, pathophysiology, clinical presentation and diagnostic approaches necessary for early detection.
Keywords
Acid Sphingomyelinase; Hepatosplenomegaly; Lysosomal storage disorder; Lipid accumulation; Neurodegeneration
Download this article as:| Copy the following to cite this article: Das S, Fiha F. S, Tanjid I. H, Datta R, Mohajon D, Dash P. R. Understanding Acid Sphingomyelinase Deficiency from a Clinical, Molecular, and Therapeutic Perspective. Biomed Pharmacol J 2026;19(2). |
| Copy the following to cite this URL: Das S, Fiha F. S, Tanjid I. H, Datta R, Mohajon D, Dash P. R. Understanding Acid Sphingomyelinase Deficiency from a Clinical, Molecular, and Therapeutic Perspective. Biomed Pharmacol J 2026;19(2). Available from: https://bit.ly/4uhOEt3 |
Introduction
Acid sphingomyelinase deficiency (ASMD) is a lysosomal storage disorder (LSD) caused by impaired activity of the acid sphingomyelinase (ASM) enzyme and, as a result, cellular accumulation of sphingomyelin. It was characterized by extensive tissue infiltration caused by foam cells, storage of lipids, and development of similar symptoms of pulmonary dysfunction, hepatosplenomegaly, and neurodegeneration.1The first case ever recorded of the ASMD disease was discovered in 1914 by Albert Niemann in an infant. However, the distinction of this disease from that of Gaucher disease was drawn in 1927 by Ludwig Pick after studying cases of infants suffering from rapid neurodegenerative disorders.2Later, it came to be called Niemann-Pick disease, NPD. It results from mutations that occur in the SMPD1 gene, which is responsible for ASM encoding. Lack of this enzyme causes lipid accumulation in body tissues, especially sphingomyelin, and is responsible for the phenotypes shown. There are some differences between ASMD A and B types. Thus, the outcomes and the fates of patients are different. ASMD type A is the most severe form of ASMD, because in this type, there is minimal to no ASM residual activity. Thus, this type is a fatal one. It shows symptoms at early infancy 2-4 months of age and little to no ASM residual activity, which causes severe central nervous system damage and quickly progressing systemic manifestation symptoms, primarily hepatosplenomegaly. The mortality is unavoidable and most often occurs in the first 3 years of life. ASMD B, by contrast, has an unusual age of onset, gradual progressive systemic expression of symptoms, and, overall, without neurological compromise. Hepatosplenomegaly is generally the presenting clinical feature, but eventually compromised pulmonary and liver function are also important.3,4Because of the wide spectrum of the disease’s severity, the lifespan of patients is highly variable, ranging from very early death to long burdened lives. In 1934, the lipid stored in this disorder was described as sphingomyelin, but not until 1966, when human-sphingomyelin-cleaving enzyme deficiency was reported, was a trial performed an NPDpatient. Characteristics of the subtypes showing the clinical spectrum of ASMD are described in Table 1.5
Table 1: Clinical spectrum of acid sphingomyelinase deficiency (ASMD) subtypes.
| Features | Severe (Neurovisceral infantile, NPD A) | Intermediate (Chronic neurovisceral, NPD A/B or variant B) | Mild (Chronic visceral NPD B) |
| Typical onset | Symptoms show up early, usually in the first few months of life. | Usually starts in childhood. | Can begin in childhood, but sometimes not until adulthood. |
| Disease course | The disease moves fast. Kids lose brain function quickly and get sicker over time. | Progresses at a moderate pace. Neurological symptoms develop, but survival is longer than in infants. | The disease creeps along slowly. Most problems are with organs, not the brain. |
| Key early features | Big liver and spleen often by 2–4 months, weak muscle tone, trouble feeding, delayed development by 6–12 months, eye problems. | Nerve problems come on later, but kids live longer than with the infantile form. | Symptoms vary, but organs get involved—people don’t usually lose brain function. |
| Neurological involvement | Severe, ongoing brain degeneration. | Symptoms vary: some have mild muscle weakness, less reflexes, problems with thinking or movement. | Hardly any or none at all. |
| Systemic manifestation | Kids don’t gain weight well, often get chest infections, and can have trouble with food going down the wrong way. | Many body systems get involved, like in the mild form. | Big liver and spleen, abnormal blood fats, slow growth or puberty, bone problems. |
| Major complications | Breathing failure, infections, and brain degeneration. | Nerve and organ problems keep getting worse. | Low platelets, lung disease, liver scarring or cirrhosis. |
| Prognosis/life expectancy | Most children don’t live past age 3. | Life span varies—a person can reach their teens or even adulthood. | Life expectancy is often normal, unless organ problems get serious. |
Although there are currently no approved medications for ASMD, ERT(Enzymatic Replacement Therapy)combined with a recombinant human ASM called olipudasealfais being developed clinically to treat ASMD’s non-neurologic symptoms.6To improve patient quality of life and decrease morbidity and disease consequences, contemporary ASMD care aims to mitigate the burden of symptoms using therapies, interventions, and lifestyle changes. People with ASMD have often received medical care from metabolic disease experts. Nevertheless, a group approach to patient care would probably include primary care physicians and additional specialists (such as pediatricians, cardiologists, pulmonologists, hepatologists, and hematologists? Due to the liver’s severe impairment in the chronic forms of ASMD, hepatologists or gastroenterologists may initially see children with chronic visceral and chronic neurovisceral ASMD, while patients with the chronic visceral form cannot show symptoms until they arrive in late adulthood because of respiratory conditions. To make sure that doctors are knowledgeable of the regular evaluations needed to manage the multisystem consequences of ASMD, it is crucial that doctors participating in patient management consult and communicate with one another.5In this review, we summarize the current knowledge on acid sphingomyelinase deficiency, including its epidemiology, pathogenesis, and pathophysiology, outlining its various disease types with their distinctive features. We discuss cellular changes in ASMD causing hepatic, pulmonary and cardiovascular diseases and their impact on the immune system. We further discuss the clinical presentation, diagnostic approaches necessary for early detection, and the available care strategies with the purpose of improving outcomes in patients. Finally, we address aspects of disease management, therapeutic developments, and factors affecting morbidity and mortality in ASMD.
Epidemiology of ASMD
ASMD affects both male and female genders, but the precise dominance is not known. Globally, about 4 to 6 babies out of every 1 million are born with ASMD and around 1 in every 250,000 people are affected. These numbers underestimate the actual impact of ASMD since there are many undiagnosed cases worldwide. So many people aren’t even aware of this condition, and this lack of knowledge often causes severe issues.7Additionally, the incidence of ASMD varies by ethnicity. The highest birth prevalence is found in the Ashkenazi Jewish population, with an estimated 2 to 3 cases per 100,000 births for NPD-A. Geographical differences have also been seen. In South America, the occurrence is about 1 in every 500,000 live births. Compared to Europe, the incidence is 1 per 250,000 live births.8The age at which ASMD occurs depends on its subtype, and the disease occurs slightly more in females. Rare genetic disorders are known to be more common among the Arab population compared to other regions worldwide. Earlier studies recommended that the high consanguinity rate, reaching up to 60%, is a huge factor contributing to the relatively high occurrence of genetic disorders in the Arab world.9There are only a few studies that have shown the incidence and prevalence of ASMD in the Arab nations. However, they expressed a relatively higher chance of ASMD compared to other regions. Moammar et al.10conducted a retrospective analysis of data from 165,130 live births in the Eastern region of Saudi Arabia for over 25 years. Eight cases of NPD type A were recorded, corresponding to an occurrencerate of 5 per 100,000 live births. The overall phenomenon of LSDs was reported as 44 per 100,000 live births. Another report from the same region time period from 1983 to 2016, reported a prevalence of NPD type A/B at 3.31 per 100,000 live births. In a 2021 systematic report of genetic disorders in Tunisia, the prevalence of ASMD type B was determined to be 1 per 200,000 live births. It is thus seen from the studies that the incidences of ASMD in the Arab countries are higher compared to the Western world.Understanding the demographic distribution or epidemiology of ASMD highlights the need to explore its underlying genetic and molecular mechanisms.11The need to investigate the underlying genetic and molecular mechanisms of ASMD is highlighted by knowledge of its prevalence and demographic distribution.
Pathogenesis of ASMD and Associated Genetic Variants
 |
Flow chart 1: Pathogenesis of Acid Sphingomyelinase Deficiency Syndrome.Click here to view Chart |
More than 720 different pathogenic variants of the SMPD1 gene associated with ASMD have been identified. These include frameshift, missense, nonsense mutations, and frame deletions. All these variations differ in geographical regions12 and the genotype-phenotype correlation is distinguished for some variants.13 To elaborate, three identified variants in the SMPD1 gene (R496L, L302P, and fsP330) are actively present in Ashkenazi Jewish ancestry and express over 90% of the ASMD patients in this population. The three variants are associated with ASMD phenotype A. The SMPD1 variant (A359D) in Chilean patients was connected with ASMD phenotype BandASMD phenotype A/B is related to p.W393G and p.Q294K genetic variations.14These genetic variations in ASMD lead to biochemical abnormalities that drive the pathophysiological changes observed in patients.
Pathophysiology of ASMD
ASMD (Acid Sphingomyelinase Deficiency) is a rare disease where the fat accumulates in the lysosome and causes disruption in the functions of cells in many organs. The actual process of this disease is not fully known, but it originates in lysosomes, which break down and recycle cellular substances. When lysosomes do not function properly, cellular processes are affected.15,16This condition accelerates the buildup of other lipids, most prominently cholesterol, ceramide, sphingosine, Lys sphingomyelin, glycosphingolipids, and bis(monoacylglycerol)phosphate. The disease primarily starts with the accumulation of a fat called sphingomyelin in lysosomes but as the fats begin to shift to the cell membrane and other regions of the cell, they lead to many secondary issues. Some of them are:
Broken cellular signaling in the membranecauses inflammation and cell death.
Problems in the synthesis of energy due to mitochondrial malfunction.
Restriction in nuclear transport.17
Lysosomes are present in many cells, except mature red blood cells. Thus, ASMD affects most cells in the body. Lysosome-dependent immune cells, like monocytes and macrophages are mostly affected. This disease affects organs like the liver and spleen, which contain a largernumber of fat-filled immune cells, and the lungs, where these cells accumulate in air sacs and surrounding tissues. Other affected organs include the heart, bones, lymphatic system, and blood-forming organs. In some cases, the disease affects the central nervous system (CNS) as fat deposits are present in glial cells and nerve cells (neurons).18ASMD has been divided into multiple subtypes as a result of these pathophysiological changes showing up clinically in different ways.
Presentations and Subtypes
Type A
Type A is the severe condition of ASMD, also known as infantile neurovisceral ASMD. The activity of the ASM enzyme in NPD-A is so low that it can be considered as non-existent.6 Such conditions during the early stages of life include growth failure, dysphagia, muscle weakness, loss of neurons, spleen and liver enlargement and lung problems. However, there is no cardiac or musculoskeletal involvement. Depending on the intensity of the disease, the development of psychomotor may normally go on for a long time after birth, but only until a maximum of between 6 and 15 months,after which there is stagnation and then backward developmental progress. Generally, drowning takes place within 3 years and is attributed to chest asphyxia induced by exposure.19
Type A/B
NPD A/B, also famous as chronic neurovisceral ASMD, is a type with slower neurological progression and longer survival.20,21 It is the mildest form of ASMD that originates in the childhood, characterized by disturbances in learning and psychiatric problems, disturbances in movements, peripheral nerve damage, and loss of coordination.22 Besides, it causes macular halo, diarrhea, abnormal liver function tests, portal hypertension, liver fibrosis and enlargement of the liver and spleen.19 Cardiac and pulmonary features reported are early-onset coronary artery disease, mixed dyslipidemia, cardiac valve disease, interstitial lung disease, abnormal pulmonary function tests, and radiological findings. Growth restriction, delayed bone maturation, reduced bone density, and bone and joint pain are common in this type.23 Deaths may occur from respiratory and liver disease during both childhood and adult life.24,25
Type B
NPD-B, also known as chronic visceral ASMD, can develop at any point from infancy to adulthood and is characterized by the slow progression of multisystem disease symptoms without neurological degeneration.26,27 NPD-B is linked to an average life expectancy, but complications such as bleeding, liver failure and respiratory failure can cause premature death.24,25 This type shows many symptoms in early childhood. Delayed puberty, fatigue, bone pain, osteopenia, thrombocytopenia and anemia are frequently observed symptoms in children.23Early in the course of the disease, mixed dyslipidemia is identified, and some individuals may even develop coronary artery disease. Mild to severe hepatic fibrosis is common in this type and the worsening of hepatic disease can cause premature death in some cases.28,29Examining the cellular alterations that take place in impacted tissues is crucial to comprehending these clinical manifestations.
Cellular Alterations
Typically, this ASM enzyme plays a role in activity that maintains cell equilibrium through cleaving and resuming membranes because it acts upon its substrate, Sphingomyelin which is part of the cellular membrane. During degradation processes, it further generates the chemical products phosphocholine and the necessary signaling lipids, Ceramides.29The deficiency of the ASM enzyme in ASMD patients causes the accumulation of sphingomyelin and hence the production of foam cells, which are lipid-laden cells that are often macrophages but can potentially impact other organ-specific cells. Indeed, the lungs, spleen (which in patients with ASMD may occasionally grow up to 10 times its normal size), bone marrow, lymph nodes, and liver all contain these foamy cells.30 Rarely, the mucosal and submucosal layers of the small and large intestines may contain them. These fat-filled cells eventually stop working correctly and die, which leads to a variety of clinical signs.31 ASMD is characterized by progressive liver disease, which is caused by sphingomyelin accumulations in hepatocytes. Similarly, splenomegaly seen in many patients is a result of lipid-laden macrophages.
HepaticDisease
ASMD is an attack on major organs, and the liver is one of them. Hepatomegaly represents the enlargement of the liver due to ASMD. Hepatomegaly, one of the most common features of ASMD, results from the buildup of sphingomyelin-filled macrophages (Kupffer cells) within the sinusoidal spaces of the liver. Such as macrophages, often called foam cells, together with the accumulation of sphingomyelin in lysosomes within hepatocytes, are responsible for liver dysfunction. For patients with the chronic visceral form of the disease, liver damage is a leading cause of death.24,25,32 According to a study that looked at the causes of mortality for 85 people with chronic ASMD, liver disease was as common among those who passed away as children and adults. This supports the idea that liver illness, at any age, was a major cause of patient mortality.25 The risk of cirrhosis, portal hypertension, and variceal hemorrhage in adults with chronic ASMD is still not well understood. Nearly all adult patients with ASMD (n = 17) showed liver fibrosis of some degree, ranging from severe cirrhosis to early-stage fibrosis, according to a phase 1 clinical trial screening.33Therefore, every safety measure needs to be followed, and one should be aware of them so that the risks can be avoided.
Splenomegaly
The most common early clinical sign of chronic ASMD is organ enlargement (organomegaly). It happens in early childhood but in milder cases, splenomegaly (enlargement of the spleen) may not be noticeable.32Upon study, almost every patient suffering from chronic ASMD experienced splenomegaly.25,26,34 Splenomegaly is caused by the lipid-filled macrophages. However, a more rapid increase in spleen size should prompt consideration of portal hypertension as a contributing factor. The enlargement of the spleen can become severe, with its volume exceeding 20 times the normal size. This can lead to hypersplenism, increasing the risks of bleeding and splenic rupture, particularly when accompanied by a decline in platelet counts.35
Pulmonary Disease
The pulmonary involvement is the most important one amongst the multisystem manifestations in sphingomyelin storage disorder.36 Radiographic evidence of spreading lung disease has been recorded in more than 90% of patients suffering from chronic visceral ASMD. According to a pathophysiological process, lipid-laden macrophages penetrate the pleura, bronchial walls, and alveolar septa in pulmonary illness, resulting in increasing lung volume restriction and impaired gas exchange.
Furthermore, as demonstrated by the ASM-deficient mice model, an inflammatory response has been observed that promotes the recruitment of macrophages to the tissues.35 There have been reports of isolated lung involvement in certain adult ASMD patients without organomegaly.37
Assessments have been conducted and are usually carried out yearly or based on symptoms, considering their severity, through pulmonary function studies, including mainly lung diffusing capacity, oxygen saturation, exercise tolerance as part of pulmonary function tests (using the modified MRC dyspnea scale) and a six-minute walk, chest X-ray even when patients do not frankly present symptoms; classically show a pattern of reticulonodular infiltration.38 Of note, imaging evidence of lung involvement correlates poorly with the severity of pulmonary impairment assessed by pulmonary function tests. Imaging for disease progression, possible with periodic chest X-ray or low-radiation high-resolution CT (HRCT) scanning every 2-4 years, allows for the timely detection of early changes and thus intervention.17
Cardiovascular Disease
Cardiac impairment is an ordinary sign observed in the patients with severe ASMD. It can also result from pulmonary disease. Overall, this problem is observed at an early age, and children are at high risk. Thus, they should be monitored and treated well. Cardiac valve abnormalities34 are commonly encountered, varying from trivial alterations with little or no clinical impact to major diseases such as mitral regurgitation and aortic stenosis, both of which have been listed in children and adults.Serial electrocardiograms annually and echocardiograms every 2–4 years can track valvular defects. Cardiac dysfunction, like coronary artery stenosis due to medial and intimal smooth muscle cell hypertrophy—presumably secondary to lysosomes containing sphingomyelin rather than pulmonary disease—has occurred in adults with longstanding ASMD.39 A baseline coronary artery study by HRCT or cardiac CT for coronary mineralization scoring at 18 years of age, and then every 2–4 years. These studies should be synchronized with pulmonary evaluations. Standard management for valve insufficiency is pharmacological treatment and/or surgical correction, which could be in the form of valve repair or valve replacement, depending on the severity of the lesion. In instances of cardiovascular diseases, interventions could include stenting or CABG, where necessary. Any surgery needs to be weighed against all potential risks that may include bleeding complications or other contraindications.5
Neurological and Ophthalmological Findings
Accordingto McGovern et al.34 all patients with NPD-B had usual mental growth, synchronization, muscle power, sensation, deep tendon reflexes, and cranial nerve function. Peripheral neuropathy was observed in 11% of cases, cherry-red spots were present in 25%, and cognitive impairment was present in 8.4%. In another statement by McGovern et al.24 neurological diseases, including sensory dyspraxia, learning disabilities, petit mal seizures, and ataxia, were documented in 12.6% of NPD-B patients. Cherry-red spots were found in one out of four NPD-A patients and one out of 21 NPD-B patients in the study done by Hollak et al. Additionally, they discovered that extreme neurological problems resulted in early death in 4 out of 5 patients suffering from NPD-A. Among patients affected with NPD-B, 4 out of 21 had neurological complications, such as intellectual disability, Parkinson’s disease, facial nerve paralysis, multiple sclerosis, and delayed motor skill development.26 It was reported that individuals with NPD-A/B had significantly more neurologic and ocular involvement than patients with NPD-B (68.8% vs. 33.3%; p = 0.032 and 55.6% vs. 15.7%; p > 0.0001, correspondingly). Additionally, neurological symptoms were more severe in patients who were 18 years of age or younger at the time of death or liver transplantation (p = 0.048).25ASMD affects different immune cell populations and affects immune function in addition to organ-specific pathology.
Acid Sphingomyelinase’s role in the immune system
Receptor binding and signaling are necessary to activate immune cells and then these activated cells identify pathogens. For the activation of the receptor, membrane platforms rich in ceramide are essential. Acid sphingomyelinase enzyme breaks down a substance called sphingomyelin and helps to create these ceramide-rich areas in the membrane. These regions bring the receptors together by clustering them in the cell membrane. This makes it easier for the receptors to activate properly and enables strong signal transmission inside the cell.40
Under a normal physiological state, ASM resides in the lysosomes. Though, under strain, under strain-for example, by the presence of pathogens and cytokines-this enzyme is released and acts on the outer membrane. It hydrolyzes the sphingomyelin present in the outer leaflet of the membrane and, after being converted to ceramide, it leads to the reorganization of standard membrane rafts into larger structures.41,42 Then in these larger structures the downstream signaling through the activity of ceramide takes place. Besides forming ceramide-enriched platforms ASM precisely controls the function of Natural Killer T cells.43 The regulation of iNKT cell activity by ASM was shown to be due to the breakdown of sphingomyelin, which has recently been identified as an inhibitor of iNKT cell activation. In this respect, this section summarizes the current concept of ASM expression in several types of immune cells. Major actions of ASM on these cells are outlined in Table 1.44 For a conception of the multiple roles of ASM outside the immunological arena, the reader is referred to three recent review articles by Henry et al. (2013), Park et al. (2020), and Perrotta et al. (2015).Macrophages impact different immune cell populations and are essential for lipid accumulation and immune function.
Macrophages
ASMD is directly linked with thedysfunction of macrophages because these cells are one of the main targets for this disorder.45,46 Macrophages of ASM KO mice show improper endocytosis of the ASM enzyme by M-6-P receptors.45 ASM is also involved in the induction or enhancement of inflammatory responses. One of the most important examples among them is the role of ASM to increase the pro-inflammatory cytokine IL-6 upon treatment with LPS and palmitic acid.47All these findings were verified by the administration of SMA-7, an ASM inhibitor, and this resulted in a remarkable reduction in ceramide formation and the release of pro-inflammatory mediators. ASMD is also crucial in the fusion of late phagosomes with lysosomes, with the facilitation of efficient delivery of lysosomal antibacterial agents and of effective cellular defense mechanisms.48 Indeed, this explains why ASM KO mice are more susceptible to infection by intracellular pathogens such as Leishmania donovani49 and Listeria monocytogenes49. By the generation of ceramide, ASM has also been associated with macrophage apoptosis induction. This was evidenced, for example, where oxidized low-density lipoproteins (LDL) induced macrophage apoptosis50,51 and in macrophage apoptosis induced by Pseudomonas aeruginosa.52 Like most pathogens, Pseudomonas aeruginosa induces macrophage killing. P. aeruginosa-induced alveolar macrophage cell death was controlled by a redox-dependent signaling cascade in which ASM was a pivotal player by generating ceramide-rich membrane platforms. These platforms are required for P. aeruginosa to induce NADPH oxidase activation to produce ROS. The ROS in turn activates ASM and ceramide-enriched platform formation that causes cell death via the JNK-dependent pathway.52 Activated macrophages secrete proteins in ASMD plasma, like chitotriosidase, that may also serve as critical clinical biomarkers.53 Chitotriosidase levels are high in ASMD but decrease sharply with treatment.54
NK Cells
NK cells are an integral part of immunity. Thus, ASM interferes with NK cell functions through CD161. ASM may work by interacting with the intracellular tail of the CD161 molecule when NK cells are activated. In fact, research showing that cross-linking CD161 with anti-CD161 antibodies caused the recruitment and activation of ASM, resulting in ceramide, validated this connection. PKB/Akt and Rsk1/MAPKAP kinase 1 are two NK cell signaling pathways that are activated by ceramide, a second messenger. Inhibition of ASM by imipramine blocks these pathways and disrupts the co-stimulatory function of CD161, impairing cell proliferation and co-stimulation of IFN-γ.55
Table 2: Functional effects of acid sphingomyelinase (ASM) on Different Cell Types.56
| Cell Type | Effects of ASM |
| Macrophages | ASM ramps up cytokine production and drives inflammatory signals. It helps phagosomes fuse with lysosomes and keeps apoptosis in check. |
| NK Cells | Affects how these cells work through CD161. causes the signaling pathways of NK cells to become active and include in NK cell apoptosis. |
| B Cells | B cell activation is facilitated by medications that cluster CD40. involved in the restoration of damaged plasma membranes. Vital to autophagic activity. |
| CD4+ T Cells | Engaged in activation controlled by TCRs. Engaged in the polarization of Th1, Th2, and Th17 subtypes. Serves as a Treg negative moderator. |
| CD8+ T Cells | ASM keeps the cell membrane flexible, making sure cytotoxic granules get released smoothly so these cells can do their job and kill targets. |
| iNKT Cells | Engaged in the activation and growth of iNKT cells. Sphingomyelin, an ASM substrate, reduces iNKT activation by blocking CD1d access to antigenic lipids. |
ASM appears to be involved in NK-organized cell death in addition to its function in macrophage apoptosis. Ceramide production in a human NK cell line is increased by IL-2 deprivation, which is ASM-dependent and causes apoptosis.57 It is one that is distinguished by the breakdown of the X-linked inhibitor of apoptosis protein through ceramide-mediated activation of cytosolic cathepsin B (CTSB), which triggers caspase-mediated apoptosis.58 IL-2, however, triggers NK cell survival through the inhibition of ASM, increasing glucosyl ceramide synthase activity and hence sphingomyelin and ceramide concentrations.57
B Cells
One of the two major branches of adaptive immunity is made up of B cells. They oversee immunity mediated by antibodies. The cells can process antigens and present them to other immune cells since they can also deliver antigens. ASM, a crucial component needed for CD40 clustering, has an impact on antigen-presenting cells.59Upon ligation of CD40, ASM moves to the exofacial leaflet of the plasma membrane and secretes ceramide that in turn induces clustering of CD40. In addition, ASM has also been shown to participate in Ca²⁺-dependent repair of plasma membrane damage in B cells. Indeed, studies have shown that inhibition or downregulation of ASM leads to defective membrane repair, whereas administration of extracellular sphingomyelinase restores and even stimulates this healing.60 B cells in a cellular model of ASMD B have defects in autophagy, mitochondrial clearance, and lipophagy. In a B lymphocyte cell line derived from a patient with ASMD B, the authors saw the occurrence of mitochondrial fragments in partially degraded structures and an atypical autophagic pattern with an extremely enhanced level of autophagic vacuoles, which was interpreted to be an autophagic stress marker.61 The outcome of such cell defects is also increased peroxidized lipid droplet content and higher cellular ROS levels through the initial and terminal stages of compromised autophagy. Canonico et al therefore went on to explore the effect that rapamycin, an inducible agent causing autophagy, would exert on such cells.60 Indeed, they succeeded in showing within this paper that rapamycin suppressed mitochondrial and intracellular ROS, mitophagy vesicle formation, hence reducing lipid droplet accumulation and improving the viability of these cells. These findings prompted the authors to propose a therapeutic use of rapamycin in LSDs, including ASMD B, by at least partial restoration of the autophagic imbalance.61
CD4+ T cells
CD4+ T cells are a subpopulation of lymphocytes that secrete particular cytokines to regulate the immune response. They have the capacity to differentiate into several different effector subtypes, each with distinct functions. These include Th1, Th2, Th17, and Treg cells, among others influenced by ASM activity.62 CD4+ T cells participate in various regulatory functions in the course of immune response regulation: these include the activation of various cells, such as B lymphocytes, cytotoxic T cells, and non-immune cells; inhibition of immune responses mainly by a subpopulation of T-regulatory (Treg) cells. Their maintenance and function depend on CD28 signaling, which has also been found to activate the ASM system.63 Specifically, how ASM directly impacts CD4+ T-cell function remains undetermined. Very recently, new research has investigated such interactions in vivo and looked into their consequences.
It binds to intracellular domains of CD3 and CD28, and this binding regulates the signaling of the receptors. The impacts are seen in naïve and memory CD4+ T cells. Further, ASM inhibition was also found to impair polarization of CD4+ T cells to Th1, Th2, and Th17 subsets, respectively, as evidenced by defective production of IFN-γ, IL-4, and IL-17.64 This was also supported by the same observation on ASM’s function in augmenting T-cell activation using a mouse model overproducing ASM exclusively in T cells. In these mice, In addition, forced expression of ASM in T cells facilitated naïve T-cell differentiation into IFN-γ-producing Th1 cells, further supporting the role of ASM in T-cell activation and polarization.65Recent research has investigated the involvement of ASM in the development of asthma in ASM knockout mice. These mice had a higher number of CD4+ T cells in the bronchoalveolar fluid, but lower production of the Th2 cytokine IL-4. This was expected, as in an ovalbumin-induced asthma model, ASM knockout mice were resistant to airway hyperresponsiveness because of a decreased Th2 response.66 This protective effect in asthma may be a result of impaired secretory pathway function in ASM-deficient mice. Lack of asm activity in enteric pathogen Citrobacter rodentium-infected mice leads to augmented infection. pathology of colitis with high Th1 and Th17 cell levels in the intestinal lamina propria.67
ASM KO mice have a significantly higher regularity of splenic Tregs.68ASM acts as a negative regulator of both the natural and induced Tregs. Similarly, in vivo ASM inhibition has been linked to a lower absolute number of conventional CD4+ T cells in the study, suggesting that ASM has further effects on CD4+ T-cell populations.63In ASM-deficient mice, effector memory CD4+ T cells show a loss of partial resistance to glucocorticoid-induced cell death. This is due to decreased IL-2 production by CD4+ T cells. However, this effect is reversed by restoring IL-2 levels, which reiterates the importance of IL-2 in the survival of CD4+ T cells during glucocorticoid treatment.69
CD8+ T Cells
Naive antigen-specific CD8+ T cells become cytotoxic T lymphocytes upon antigen recognition. CTLs contain lytic granules whose contents are released upon TCR engagement of the appropriate antigen on APCs. The granules contain granzyme A and B and perforin that form pores in the target cell membrane so that granzymes may penetrate the cytoplasm and induce programmed cell death by activating caspases.
ASM plays a role in CTL activation by facilitating granule release. In ASM-deficient mice, cytotoxic granule release is delayed, leading to delayed lymphocytic choriomeningitis virus clearance.70,71 However, as compared to wild-type mice, the mRNA, protein, and enzymatic processes of granzyme A, granzyme B, and perforin remain unchanged. Both mouse types exhibit normal CD8+ T cell growth and antigen-specific activation. Indeed, in ASM-deficient mice, larger granzyme-positive granule clusters accumulate closer to the immunological synapse. In the absence of ASM, vesicle size shrinks only 44% following fusion with the plasma membrane, resulting in poorly released cytotoxic molecules. These findings were further supported by the pharmacological inhibition of ASM activity in CTLs.71 These findings emphasize the role of ASM in the release of cytolytic granules from CTLs and suggest the possible application of ASM inhibitors in the therapy of immunopathological conditions mediated by excessive cytolytic activities. Indeed, ASM inhibition was clinically explored for the therapeutic benefits in patients suffering from cystic fibrosis against infections of the lung by bacteria.72
iNKT Cells
iNKT cells are T cells responding to lipids by a limited repertoire of TCR, which is specific to the MHC class-I-like molecule CD1d. iNKT cell activity is controlled by non-self and self-lipid ligands denoted by CD1d. iNKT cells are also in charge of tumor growth regulation and infection.73 Although there is a balance between antigenic lipids and non-antigenic lipids that are involved in CD1d in the regulation of iNKT cell activation, little is known about the functional applicability of such non-antigenic lipids that can potentially suppress the activation of the CD1d-restricted NKT cells.74,75 ASM KO mice and cells from patients without ASM were used in the study. Mouse deficiency of ASM decreased CD1d-restricted presentation of both endogenous and exogenous lipid antigens to the iNKT. This dysfunction hampered the development of iNKT in the thymus further leading to low levels both in the thymus and systemically. A schematic outline for such a suggested mechanism is given in Figure 1. Indeed, such alterations were improvedin young ASM KO miceby the ASM replacement therapy.43 The effectiveness of ASM in ASM KO mice certainly instills faith in the therapeutic potential of ASM therapy for ASMD patients, particularly in the case of pulmonary infections. Lung infections are the most common secondary symptoms of the disease, and iNKT cells play an important role in antimicrobial immunity against the common respiratory pathogens such as Pneumococcus and Pseudomonas.76
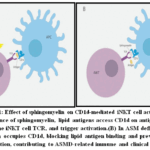 |
Figure 1: Effect of sphingomyelin on CD1d-mediated iNKT cell activation. |
Clinical Overview and Diagnosis
Clinical signs are the main clues that point towards the presence of ASMD. Niemann-Pick C, Gaucher disease, acid lipase insufficiency, and other LSDs associated with distinctive hepatosplenomegaly can also exhibit these signs. In this regard, the diagnosis of ASMD requires insufficient ASM enzymatic activity which can be determined by using detached leukocytes, dried blood spots, or cultured skin fibroblasts.77 A genetic test for the SMPD1 gene confirms the ASMD diagnosis and enables the patient to get genetic counseling after the diagnosis has been confirmed.78
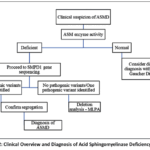 |
Flow chart 2: Clinical Overview and Diagnosis of Acid Sphingomyelinase Deficiency Syndrome. |
Diagnostic Approaches
Enzyme activity testing in ASMD
If there is any doubt of ASMD, the first thing that should be done is an enzyme assay for ASM function and then the gene sequencing after confirming the biochemical diagnosis.77 To distinguish ASMD from Gaucher disease, glucocerebrosidase activity should be measured at the same time.79The diagnosis of this disease should be confirmed by explaining reduced enzyme activity below the standard cut-off range, while accounting for the many distinct genetic variants of uncertain significance.80 Other supportive clinical and laboratory features include lipid-laden foam cells and mixed dyslipidemia, which are highly suggestive of ASMD but do not replace the confirmation by enzyme testing. A molecular test for known pathogenic or familial mutations can confirm the diagnosis in case there is a strong suspicion of ASMD based on clinical findings. However, in cases where molecular studies reveal variants of uncertain significance, confirmation needs to be made through an enzyme assay.
Whole blood is usually required for laboratories that sample from all over the world, so that the same sample can be used for second-tier testing as well 5. DBS testing has limits, including the impact of anemia and recent transfusions on the results, although it can be utilized for particularly stable DBS.81 Due to requirements for radiochemical licenses, MS/MS is the preferred procedure for analyzing ASM activity over fluorescence-based assays and radio-labeled native sphingomyelin substrate because of its higher extended measurement range with better accuracy measurement of enzyme activity at the lowest detectable levels.82Treatment plans targeted at lowering morbidity and mortality in ASMD patients are informed by an accurate diagnosis.
General Laboratory Tests
Blood Counts: The ASMD patients more often than not show mild thrombocytopenia which is already suggested by routine blood tests and anemia along with leucopenia or neutropenia may be present.
Lipid profile: People with ASMD have highly aberrant lipid profiles. The majority of the anomalies involve decreased HDL cholesterol and increased triglyceride and total cholesterol levels.83
Imaging: The painting of the liver and enlargement of the spleen are usually detected on simple radiography or during physical examination. However, to assess the size accurately an echogenicity ultrasound is better. CT scans and MRI are able to provide more confidence for these assessments. Interstitial lung disease may also be suggested by the images taken by chest X-rays or CT images.84
Table 3: Laboratory diagnostic workflow for acid sphingomyelinase deficiency (ASMD).
This table organizes the steps and actions related to diagnosing and confirming ASMD based on the provided content.
| Step | Key Purpose | Main Action |
| Clinical Suspicion | Identify patients requiring evaluation. | Assess symptoms and clinical features suggestive of ASMD |
| Biomarkers Assessment | Support preliminary diagnosis | Measure disease-related biomarkers (e.g., NPS and other relevant markers) |
| Enzyme Activity Testing | Confirm biochemical deficiency | Measure acid sphingomyelinase (ASM) activity |
| Molecular Testing | Identify genetic cause | Perform gene analysis to detect pathogenic variants or variants of uncertain significance (VUS) |
| Variant Interpretation | Establish diagnostic validity | Confirm biallelic pathogenic variants consistent with disease |
| Diagnostic Confirmation | Finalize diagnosis | Integrate clinical, biochemical, and genetic findings |
| Genetic Counseling | Guide patient management | Provide counseling and family risk assessment |
Treatment and Management of ASMD
There is still no proper treatment available for ASMD. But currently, enzyme replacement therapy (ERT) using human recombinant acid sphingomyelinase olipudase alfa is in clinical development for the treatment of non-neurological ASMD manifestations. There are two clinical trials are being conducted and they are in phase 2 (NCT02004704) and 2/3 (NCT02004691).85 In addition, three studies have been completed, one in phase 1/2 (NCT02292654).86 and the other two in phase 1 (NCT00410566; NCT01722526).87,88.However, there are still some important limitations. The current ERT cannot pass the blood-brain barrier. Therefore, it is not useful for patients with central nervous system involvement. Consequently, in severe cases, the neurological symptoms are still progressive.89Moreover, long-term information on safety, immunogenicity, and durability of treatment effects is still limited, and the high cost of treatment is a major barrier, especially in developing countries.90Another significant challenge in the management of ASMD is the genetic and clinical variability of patients. Various mutations in the SMPD1 gene cause different severities of the disease, making it difficult to predict the prognosis of patients.91 Moreover, the prevalence of ASMD is low, and this makes it difficult to perform large-scale clinical trials.89 Since there is no permanent treatment approach available, the treatment of ASMD can require a combined effort of specialists working together to enhance the life of the patient. Specialists required include pediatricians, neurologists, hepatologists, ophthalmologists, and many others.92 Death is still a significant issue despite the treatment approaches available, and this highlights the need for optimization. Mortality is still a major concern despite available treatments, underscoring the necessity of optimizing care.
How is Optimal Care Delivered for ASMD
Some ASMD patients are able to cope with only a multi-system disease with mild progress in certain areas of management. Along with the healthcare experts, ASMD patients have reported a better experience only if they had been followed up by a group of multidisciplined healthcare professionals who are have the proper knowledge about ASMD. Such patients should be referred to the centers that are well recognized for ASMD management if available. Strength of Guidance: 1, Quality of Evidence: A, Specialist Viewpoint: Full agreement 75%, partial agreement 19%, slight agreement 6%, partial disagreement 0%, full disagreement 0%. Most clinical research papers about rare diseases, which are classified as multisystemic, point towards greater patientsatisfaction after utilizing specialized services. There are reports of improvement in compliance rates as well as better attendance for appointments in comprehensive care clinics compared to traditional approaches.93
If we consider the outline of the health systems, the competencies offered and the patient’s requirements of that particular country, it is possible to establish a multidisciplinary group (MDG) to help coordinate ASMD patients in an effective manner. This approach promotes sharing and coordination of care while also ensuring that barriers to communication across disciplines are removed. In order to monitor the ASMD which is progressive and multisystemic, an international expert guideline has been formulated. Monitoring goals should be set at the time of diagnosis. They should be reviewed regularly to make sure that the complications of the disease are under control, and the patient’s quality of life is enhanced.94
Table 4: Guideline-recommended teamwork for patients with acid sphingomyelinase deficiency (ASMD)17
| Discipline | Primary Role in ASMD Management | Recommended Involvement |
| Primary healthcare provider | Coordinates overall care, monitors general health and facilitates specialist referral. | All Patients. |
| Metabolic Specialist | Confirms diagnosis, oversees disease monitoring and guides treatment strategy. | All Patients. |
| Neurologist | Evaluates and manages neurological involvement. | All Patients. |
| Hepatologist | Monitors liver function and manges hepatic complications. | As clinically indicated. |
| Hematologist | Assesses hematologic abnormalities and bleeding risk. | Asclinically indicated. |
| Pulmonologist | Evaluates respiratory function and manages pulmonary disease. | Asclinically indicated. |
| Genetic counsellor | Provides family risk assessment, inheritance counseling and reprodictive guidance. | All patients. |
| Lipidologist/Cardiologist | Manages dyslipidemia and cardiovascular risk. | As clinically indicated. |
| Psychology or psychiatry specialist | Addresses emotional, behavioral and mental health needs. | As clinically indicated. |
| Speech and Communicationspecialist | Evaluates swallowing and communication difficulties; provides supportive therapy. | As clinically indicated. |
How Should Burnout of Illness be Assessed
The presentation of ASMD along with the survival rate greatly depends on the subtype of the disease, age at which it was diagnosed, the organs affected and the complications associated with it.95 Out of the 3 types, ASMD type A has the worst survival rate as it has a shorter life expectancy with a relatively consistent advancement of the disease.94,95 ASMD type B, on the opposite end of the spectrum, has a wide range of symptoms that affect the disease’s severity, progression pace, and life expectancy.4,96,97 A&B is a type where the patients have combined traits of both type A and B and this mix goes hand in hand with a slower progression of neurodegeneration. There exist clinical guidelines to help monitor ASMD.98 and more importantly, there exists a marking triad that can help in the diagnosis detection early stages, and help in changing the course of treatment. The key triad that marks ASMD amalgamates an abnormal methylation pattern of TP73, increased mRNA levels of CPHM, and diminished levels of CD11b-expressing monocytes (Table 4).
Table 5: Recommended ClinicalMonitoring andAssessments for patients withASMD
| Assessment Domain | Clinical Purpose | Frequency | Applicability |
| Clinical history | Determine the disease’s natural history. | At diagnosis and routine follow up | All patients |
| Physical check-up | At least once a year, record growth parameters. | Upon diagnosis, and then at 6–12-month intervals or during each visit. | Asclinically indicated. |
| Nutritional assessment | Assessment of nutritional status. | Initially upon diagnosis, followed by assessment at each clinical visit. | All Patients. |
| Respiratory assessment | Evaluate recurring chest infections. | Following diagnosis, you should conduct assessments at each follow-up appointment. | All Patients. |
| Musculoskeletal assessment | Check for discomfort in the extremities and/or fractures. | Every appointment after the diagnosis. | All Patients. |
| Neurological assessment | Assess neurological function carefully. | At diagnosis and as clinically indicated | As clinically indicated. |
| Laboratory testing | Serum tests including albumin, coagulation matrices, and liver enzyme plus transaminases (ALT and AST) help analyze the liver. | Initially at diagnosis, with subsequent assessments conducted at least yearly. | As clinically indicated. |
| Diagnostic imaging | Lipid profile measurement.When necessary, radiologic measurements of the size of the liver and spleen. | Initially at diagnosis, then repeated as needed. | As clinically indicated. |
| Swallowing and feeding assessment | Identify dysphagia and aspiration risk. | Baseline and periodic reassessment based on symptoms. | As clinically indicated. |
| Developmental or cognitive assessment | Monitoring educational requirements and developmental progress through developmental assessment. | Performed after the diagnosis and then at each clinical visit. | As clinically indicated. |
| Neuropsychiatric evaluation | Record psychiatric symptoms and track treatment outcomes. | Initially at diagnosis, then repeated every 6 to 12 months as needed. | As clinically indicated. |
| Family support and assistance | At every visit, determine the resources and support needs of the family. | Every appointment after the diagnosis. | As clinically indicated. |
Mortality
In the analysisof Wasserstein et al three patients, aged 9 to 18 years, passed away due to liver failure, traumatic subdural hematoma, and long-term difficulties due to an unsuccessful bone marrow transplant. Four patients suffering from NPD-A lost their lives because of extreme neurological manifestations, subdural hematoma, malignant edema, and pneumonia, according to Hollak et al.96 Five patients with NPD-B lost their lives from this illness. Three of these were caused by worsening lung disease. One patient died from cancer, and the cause of death is unknown for another patient. In an investigation by McGovern and colleagues, they discovered 18 deaths in NPD-B patients. The main causes for these were respiratory failure, which caused five deaths. Liver failure was responsible for three deaths. Another three deaths occurred due to complications after a bone marrow transplant. Other causes of death included bleeding in the brain, failure of multiple organs, heart failure, liver cancer, a burst spleen vein, and bleeding after surgery e bleeding was among the causes of mortality in one patient each.94 In a retrospective analysis of 100 patients, 12 were deceased. Among these NPD-A/B patients, ten deaths occurred at a mean age of 2.4 years. The causes included lung disorders (n = 1), respiratory failure (n = 2), pneumonia (n = 3), hepatic failure (n = 1), or unknown reasons (n = 3). Two NPD-B patients also died: one at 42 years from respiratory failure and another at 2 years from hepatic failure.99 A recent study reported the deaths of eight patients due to ASMD. Three patients, aged 20, 35, and 71, passed away due to pneumonia. Two patients, aged 16 and 65, died from liver failure and cancer. One patient, aged 14, passed away due to a splenic vein tear. The other two individuals, aged 17 and 56, died from portal hypertension with esophageal varices and multiorgan failure. Pneumonia was the primary cause of mortality in these patients.100
Conclusion
ASMD is an ultra-rare lysosomal storage disease distributed worldwide and associated with over 200 variants in the SMPD1 gene. The pathogenesis results from deficient acid sphingomyelinase activity and disrupted sphingomyelin metabolism, leading to widespread pathophysiological consequences: cellular changes that underlie the multisystem involvement include the liver, spleen, lungs, and, in severe forms, the central nervous system. It manifests as a number of fairly well-defined phenotypes, which range from chronic non-neuronopathic forms to rapid neurodegenerative variants. Besides its function in lipid metabolism, acid sphingomyelinase is also an important modulator in immune regulation but especially in controlling Natural Killer T cell activation. Diagnostic methods such as biochemical analysis, genetic analysis, and neuroimaging studies are used for early detection and disease classification. However, the limitations in sensitivity, accessibility, and standardization of these methods emphasize the need for further advances in the diagnostic area. In terms of therapy, enzyme replacement therapy is a promising area, but its failure to target the central nervous system, variable efficacy, and high cost create many problems. Taken together, these results indicate that future studies should aim at genotype-phenotype correlation analysis, CNS-targeted therapies, and standardization of diagnostic and monitoring procedures to improve patient survival and quality of life.
Acknowledgment
The authors express gratitude to the Department of Pharmacy, University of Science and Technology Chittagong.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials
Permission to reproduce material from other sources
Not Applicable
Author Contributions
- Pritesh Ranjan Dash: Supervision (lead), Project Administration(lead), Conceptualization(lead), Formal analysis (supporting).
- Swapnil Das: Supervision (equal), Project Administration(equal), Conceptualization(equal), Formal analysis (lead), Methodology(lead), Investigation(lead), Data Curation(lead).
- Fatiha Sultana Fiha: Methodology(equal), Investigation(equal), Data Curation(equal), Resources(lead), Writing – Original Draft(lead), Writing – Review & Editing(lead).
- Rohit Datta: Methodology(supportive), Investigation(supportive), Data Curation(supportive), Resources(equal), Writing – Original Draft(equal), Writing – Review & Editing(equal).
- Dip Mohajon: Resources(supportive), Visualization(lead), Data Curation(supportive).
- Irfan Hoque Tanjid: Resources(supportive), Visualization(equal), Data Curation(supportive).
References
- Pinto C, Sousa D, Ghilas V, Dardis A, Scarpa M, Macedo MF. Acid Sphingomyelinase Deficiency: A Clinical and Immunological Perspective. Int J Mol Sci 2021, Vol 22,. 2021;22(23). doi:10.3390/ijms222312870
CrossRef - Schuchman E, metabolism RDM genetics and, 2017 undefined. Types a and B Niemann-pick disease. Elsevier. Accessed September 9, 2025.https://www.sciencedirect.com/science/article/pii/ S1096719216303171
- McGovern MM, Aron A, Brodie SE, Desnick RJ, Wasserstein MP. Natural history of Type A Niemann-Pick disease. Neurology. 2006;66(2):228-232. doi:10.1212/01.WNL.0000194208.08904.0C
CrossRef - Wasserstein MP, Desnick RJ, Schuchman EH, et al. The Natural History of Type B Niemann-Pick Disease: Results From a 10-Year Longitudinal Study. Pediatrics. 2004;114(6):e672-e677. doi:10.1542/PEDS.2004-0887
CrossRef - Wasserstein M, Dionisi-Vici C, Giugliani R, et al. Recommendations for clinical monitoring of patients with acid sphingomyelinase deficiency (ASMD). Mol Genet Metab. 2019;126(2):98-105. doi:10.1016/J.YMGME.2018.11.014
CrossRef - Thurberg BL. Autopsy pathology of infantile neurovisceral ASMD (Niemann-Pick Disease type A): Clinicopathologic correlations of a case report. Mol Genet Metab Reports. 2020;24:100626. doi:10.1016/J.YMGMR.2020.100626
CrossRef - Villeneuve T, Jamme T, Schwob R, Levade T, Prévot G. Advanced strategies for detecting acid sphingomyelinase deficiency type B with attenuated phenotypes. Orphanet J Rare Dis 2025 201. 2025;20(1):252-. doi:10.1186/s13023-025-03746-9
CrossRef - Acuña M, Martínez P, Moraga C, et al. Epidemiological, clinical and biochemical characterization of the p.(Ala359Asp) SMPD1 variant causing Niemann-Pick disease type B. Eur J Hum Genet. 2016;24(2):208-213. doi:10.1038/EJHG.2015.89;TECHMETA=109,23,38,42,70,77;SUBJMETA=317,692,699;KWRD=METABOLIC+DISORDERS
CrossRef - Al-Gazali L, Hamamy H, Al-Arrayad S. Genetic disorders in the Arab world. BMJ. 2006;333(7573):831-834. doi:10.1136/BMJ.38982.704931.AE
CrossRef - Moammar H, Cheriyan G, Mathew R, Al-Sannaa N. Incidence and patterns of inborn errors of metabolism in the eastern province of saudi arabia, 1983-2008. Ann Saudi Med. 2010;30(4). doi:10.4103/0256-4947.65254;WEBSITE:WEBSITE:ASM-SITE;PAGEGROUP:STRING:PUBLICATION
CrossRef - Mezzi N, Messaoud O, Mkaouar R, et al. Spectrum of Genetic Diseases in Tunisia: Current Situation and Main Milestones Achieved. Genes 2021, Vol 12,. 2021;12(11). doi:10.3390/genes12111820
CrossRef - Mezzi N, Messaoud O, Mkaouar R, et al. Spectrum of genetic diseases in tunisia: Current situation and main milestones achieved. Genes (Basel). 2021;12(11):1820. doi:10.3390/GENES12111820/S1
CrossRef - Zhang H, Wang Y, Gong Z, et al. Identification of a distinct mutation spectrum in the SMPD1 gene of Chinese patients with acid sphingomyelinase-deficient Niemann-Pick disease. Orphanet J Rare Dis. 2013;8(1):1-8. doi:10.1186/1750-1172-8-15/TABLES/2
CrossRef - Levran O, Desnick RJ, Schuchman EH. Type A Niemann‐Pick disease: A frameshift mutation in the acid sphingomyelinase gene (fsP330) occurs in Ashkenazi Jewish patients. Hum Mutat. 1993;2(4):317-319. doi:10.1002/humu.1380020414
CrossRef - Walkley SU, Vanier MT. Secondary lipid accumulation in lysosomal disease. Biochim Biophys Acta – Mol Cell Res. 2009;1793(4):726-736. doi:10.1016/J.BBAMCR.2008.11.014
CrossRef - Breiden B, Sandhoff K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int J Mol Sci 2020, Vol 21, Page 2566. 2020;21(7):2566. doi:10.3390/IJMS21072566
CrossRef - Geberhiwot T, Wasserstein M, Wanninayake S, et al. Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann–Pick disease types A, B and A/B). Orphanet J Rare Dis 2023 181. 2023;18(1):85-. doi:10.1186/s13023-023-02686-6
CrossRef - Pinto C, Sousa D, Ghilas V, Dardis A, Scarpa M, Macedo MF. Acid Sphingomyelinase Deficiency: A Clinical and Immunological Perspective. Int J Mol Sci 2021, Vol 22,. 2021;22(23). doi:10.3390/ijms222312870
CrossRef - Floris G, Di Stefano F, Melis R, Cherchi MV, Marrosu F. Isolated bipallidal lesions caused by extrapontine myelinolysis. Neurology. 2013;81(19):1722-1723. doi:10.1212/01.WNL.0000435297.80023.9E
CrossRef - Wasserstein MP, Aron A, Brodie SE, Simonaro C, Desnick RJ, McGovern MM. Acid sphingomyelinase deficiency: Prevalence and characterization of an intermediate phenotype of Niemann-Pick disease. J Pediatr. 2006;149(4):554-559. doi:10.1016/J.JPEDS.2006.06.034
CrossRef - Pavlů-Pereira H, Asfaw B, Poupětová H, et al. Acid sphingomyelinase deficiency. Phenotype variability with prevalence of intermediate phenotype in a series of twenty-five Czech and Slovak patients. A multi-approach study. J Inherit Metab Dis. 2005;28(2):203-227. doi:10.1007/S10545-005-5671-5
CrossRef - McGovern MM, Dionisi-Vici C, Giugliani R, et al. Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet Med. 2017;19(9):967-974. doi:10.1038/GIM.2017.7
CrossRef - Wasserstein M, Godbold J, McGovern MM. Skeletal manifestations in pediatric and adult patients with Niemann Pick disease type B. J Inherit Metab Dis. 2013;36(1):123-127. doi:10.1007/S10545-012-9503-0
CrossRef - McGovern MM, Lippa N, Bagiella E, Schuchman EH, Desnick RJ, Wasserstein MP. Morbidity and mortality in type B Niemann–Pick disease. Genet Med. 2013;15(8):618-623. doi:10.1038/GIM.2013.4
CrossRef - Cassiman D, Packman S, Bembi B, et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases. Mol Genet Metab. 2016;118(3):206-213. doi:10.1016/J.YMGME.2016.05.001
CrossRef - Hollak CEM, de Sonnaville ESV, Cassiman D, et al. Acid sphingomyelinase (Asm) deficiency patients in The Netherlands and Belgium: Disease spectrum and natural course in attenuated patients. Mol Genet Metab. 2012;107(3):526-533. doi:10.1016/J.YMGME.2012.06.015
CrossRef - Wasserstein MP, Desnick RJ, Schuchman EH, et al. The Natural History of Type B Niemann-Pick Disease: Results From a 10-Year Longitudinal Study. Pediatrics. 2004;114(6):e672-e677. doi:10.1542/PEDS.2004-0887
CrossRef - Ishii H, Takahashi T, Toyono M, et al. Acid sphingomyelinase deficiency: Cardiac dysfunction and characteristic findings of the coronary arteries. J Inherit Metab Dis. 2006;29(1):232-234. doi:10.1007/s10545-006-0226-y
CrossRef - Zeidan YH, Hannun YA. The Acid Sphingomyelinase/Ceramide Pathway: Biomedical Significance and Mechanisms of Regulation. Curr Mol Med. 2010;10(5):454-466. doi:10.2174/156652410791608225
CrossRef - McGovern MM, Avetisyan R, Sanson BJ, Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis. 2017;12(1):1-13. doi:10.1186/S13023-017-0572-X/FIGURES/1
CrossRef - Sagaert X, Tousseyn T, De Hertogh G, Geboes K. Macrophage-related diseases of the gut: A pathologist’s perspective. Virchows Arch. 2012;460(6):555-567. doi:10.1007/S00428-012-1244-9/METRICS
CrossRef - McGovern MM, Avetisyan R, Sanson BJ, Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis. 2017;12(1):1-13. doi:10.1186/S13023-017-0572-X/FIGURES/1
CrossRef - Thurberg BL, Wasserstein MP, Schiano T, et al. Liver and skin histopathology in adults with acid sphingomyelinase deficiency (niemann-pick disease type B). Am J Surg Pathol. 2012;36(8):1234-1246. doi:10.1097/PAS.0B013E31825793FF
CrossRef - McGovern MM, Wasserstein MP, Giugliani R, et al. A Prospective, Cross-sectional Survey Study of the Natural History of Niemann-Pick Disease Type B. Pediatrics. 2008;122(2):e341-e349. doi:10.1542/PEDS.2007-3016
CrossRef - Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis. 2007;30(5):654-663. doi:10.1007/S10545-007-0632-9/METRICS
CrossRef - Minai OA, Sullivan EJ, Stoller JK. Pulmonary involvement in Niemann–Pick disease: Case report and literature review. Respir Med. 2000;94(12):1241-1251. doi:10.1053/RMED.2000.0942
CrossRef - Nicholson AG, Florio R, Hansell DM, et al. Pulmonary involvement by Niemann–Pick disease. A report of six cases. Histopathology. 2006;48(5):596-603. doi:10.1111/J.1365-2559.2006.02355.X
CrossRef - Mendelson DS, Wasserstein MP, Desnick RJ, et al. Type B Niemann-Pick Disease: Findings at Chest Radiography, Thin-Section CT, and Pulmonary Function Testing1. https://doi.org/101148/radiol2381041696. 2006;238(1):339-345. doi:10.1148/radiol.2381041696
CrossRef - Ishii H, Takahashi T, Toyono M, et al. Acid sphingomyelinase deficiency: Cardiac dysfunction and characteristic findings of the coronary arteries. J Inherit Metab Dis. 2006;29(1):232-234. doi:10.1007/S10545-006-0226-Y
CrossRef - Bai A, Guo Y. Acid sphingomyelinase mediates human cd4+ t-cell signaling: Potential roles in t-cell responses and diseases. Cell Death Dis. 2017;8(7):e2963-e2963. doi:10.1038/CDDIS.2017.360;SUBJMETA
CrossRef - Chung HY, Claus RA. Keep Your Friends Close, but Your Enemies Closer: Role of Acid Sphingomyelinase During Infection and Host Response. Front Med. 2021;7:616500. doi:10.3389/FMED.2020.616500/FULL
CrossRef - Tawk C, Nigro G, Lopes IR, et al. Stress‐induced host membrane remodeling protects from infection by non‐motile bacterial pathogens. EMBO J. 2018;37(23). doi:10.15252/EMBJ.201798529
CrossRef - Melum E, Jiang X, Baker KD, et al. Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin. Nat Immunol. 2019;20(12):1644-1655. doi:10.1038/S41590-019-0504-0;SUBJMETA
CrossRef - Pinto C, Sousa D, Ghilas V, Dardis A, Scarpa M, Macedo MF. Acid Sphingomyelinase Deficiency: A Clinical and Immunological Perspective. Int J Mol Sci 2021, Vol 22, Page 12870. 2021;22(23):12870. doi:10.3390/IJMS222312870
CrossRef - Aldosari MH, de Vries RP, Rodriguez LR, et al. Liposome-targeted recombinant human acid sphingomyelinase: Production, formulation, and in vitro evaluation. Eur J Pharm Biopharm. 2019;137:185-195. doi:10.1016/J.EJPB.2019.02.019
CrossRef - Borie R, Crestani B, Guyard A, Lidove O. Interstitial lung disease in lysosomal storage disorders. Eur Respir Rev. 2021;30(160). doi:10.1183/16000617.0363-2020
CrossRef - Jin J, Zhang X, Lu Z, et al. Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. https://doi.org/101152/ajpendo002512013. 2013;305(7):853-867. doi:10.1152/AJPENDO.00251.2013
CrossRef - Xiang H, Jin S, Tan F, Xu Y, Lu Y, Wu T. Physiological functions and therapeutic applications of neutral sphingomyelinase and acid sphingomyelinase. Biomed Pharmacother. 2021;139:111610. doi:10.1016/J.BIOPHA.2021.111610
CrossRef - Schramm M, Herz J, Haas A, Krönke M, Utermöhlen O. Acid sphingomyelinase is required for efficient phago-lysosomal fusion. Cell Microbiol. 2008;10(9):1839-1853. doi:10.1111/J.1462-5822.2008.01169.X
CrossRef - DEIGNER HP, CLAUS R, BONATERRA GA, et al. Ceramide induces aSMase expression: implications for oxLDI-induced apoptosis. FASEB J. 2001;15(3):807-814. doi:10.1096/FASEBJ.15.3.807
CrossRef - Zhao M, Pan W, Shi RZ, et al. Acid Sphingomyelinase Mediates Oxidized-LDL Induced Apoptosis in Macrophage via Endoplasmic Reticulum Stress. J Atheroscler Thromb. 2016;23(9):1111-1125. doi:10.5551/JAT.32383
CrossRef - Wu Y, Li C, Peng H, et al. Acid Sphingomyelinase Contributes to the Control of Mycobacterial Infection via a Signaling Cascade Leading from Reactive Oxygen Species to Cathepsin D. Cells 2020, Vol 9, Page 2406. 2020;9(11):2406. doi:10.3390/CELLS9112406
CrossRef - Diaz GA, Jones SA, Scarpa M, et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet Med. 2021;23(8):1543-1550. doi:10.1038/S41436-021-01156-3
CrossRef - Eskes ECB, Sjouke B, Vaz FM, et al. Biochemical and imaging parameters in acid sphingomyelinase deficiency: Potential utility as biomarkers. Mol Genet Metab. 2020;130(1):16-26. doi:10.1016/J.YMGME.2020.02.002
CrossRef - Pozo D, Valés-Gómez M, Mavaddat N, Williamson SC, Chisholm SE, Reyburn H. CD161 (Human NKR-P1A) Signaling in NK Cells Involves the Activation of Acid Sphingomyelinase 1. J Immunol. 2006;176:2397-2406.
CrossRef - Pinto C, Sousa D, Ghilas V, Dardis A, Scarpa M, Macedo MF. Acid Sphingomyelinase Deficiency: A Clinical and Immunological Perspective. Int J Mol Sci 2021, Vol 22,. 2021;22(23). doi:10.3390/ijms222312870
CrossRef - Taguchi Y, Kondo T, Watanabe M, et al. Interleukin-2-induced survival of natural killer (NK) cells involving phosphatidylinositol-3 kinase-dependent reduction of ceramide through acid sphingomyelinase, sphingomyelin synthase, and glucosylceramide synthase. Blood. 2004;104(10):3285-3293. doi:10.1182/BLOOD-2004-03-0900
CrossRef - Taniguchi M, Ogiso H, Takeuchi T, Kitatani K, Umehara H, Okazaki T. Lysosomal ceramide generated by acid sphingomyelinase triggers cytosolic cathepsin B-mediated degradation of X-linked inhibitor of apoptosis protein in natural killer/T lymphoma cell apoptosis. Cell Death Dis. 2015;6(4):e1717-e1717. doi:10.1038/CDDIS.2015.82;TECHMETA
CrossRef - Michaud M, Mauhin W, Villeneuve T, Lidove O. [Acid sphingomyelinase deficiency: A review]. La Rev Med interne. 2025;46(11):654-661. doi:10.1016/j.revmed.2025.03.425
CrossRef - Miller H, Castro-Gomes T, Corrotte M, et al. Lipid raft–dependent plasma membrane repair interferes with the activation of B lymphocytes. J Cell Biol. 2015;211(6):1193-1205. doi:10.1083/JCB.201505030
CrossRef - Canonico B, Cesarini E, Salucci S, et al. Defective Autophagy, Mitochondrial Clearance and Lipophagy in Niemann-Pick Type B Lymphocytes. PLoS One. 2016;11(10):e0165780. doi:10.1371/JOURNAL.PONE.0165780
CrossRef - Bai A, Guo Y. Acid sphingomyelinase mediates human cd4+ t-cell signaling: Potential roles in t-cell responses and diseases. Cell Death Dis. 2017;8(7):e2963-e2963. doi:10.1038/CDDIS.2017.360;SUBJMETA
CrossRef - Schneider-Schaulies J, Beyersdorf N. CD4 + Foxp3 + regulatory T cell-mediated immunomodulation by anti-depressants inhibiting acid sphingomyelinase. Biol Chem. 2018;399(10):1175-1182. doi:10.1515/HSZ-2018-0159/MACHINEREADABLECITATION/RIS
CrossRef - Bai A, Kokkotou E, Zheng Y, Robson SC. Role of acid sphingomyelinase bioactivity in human CD4+ T-cell activation and immune responses. Cell Death Dis. 2015;6(7):e1828-e1828. doi:10.1038/CDDIS.2015.178;TECHMETA
CrossRef - Hose M, Günther A, Abberger H, et al. T cell-specific overexpression of acid sphingomyelinase results in elevated T cell activation and reduced parasitemia during Plasmodium yoelii infection. Front Immunol. 2019;10(MAY):454364. doi:10.3389/FIMMU.2019.01225/BIBTEX
CrossRef - Böll S, Ziemann S, Ohl K, et al. Acid sphingomyelinase regulates TH2 cytokine release and bronchial asthma. Allergy. 2020;75(3):603-615. doi:10.1111/ALL.14039
CrossRef - Meiners J, Palmieri V, Klopfleisch R, et al. Intestinal acid sphingomyelinase protects from severe pathogen-driven colitis. Front Immunol. 2019;10(JUN):458492. doi:10.3389/FIMMU.2019.01386/BIBTEX
CrossRef - Zhou Y, Salker MS, Walker B, et al. Acid Sphingomyelinase (ASM) is a Negative Regulator of Regulatory T Cell (Treg) Development. Cell Physiol Biochem. 2016;39(3):985-995. doi:10.1159/000447806
CrossRef - Schweingruber N, Fischer HJ, Fischer L, et al. Chemokine-mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. 2014;127(5):713-729. doi:10.1007/S00401-014-1248-4/METRICS
CrossRef - Xiang H, Jin S, Tan F, Xu Y, Lu Y, Wu T. Physiological functions and therapeutic applications of neutral sphingomyelinase and acid sphingomyelinase. Biomed Pharmacother. 2021;139:111610. doi:10.1016/J.BIOPHA.2021.111610
CrossRef - Herz J, Pardo J, Kashkar H, et al. Acid sphingomyelinase is a key regulator of cytotoxic granule secretion by primary T lymphocytes. Nat Immunol. 2009;10(7):761-768. doi:10.1038/NI.1757;KWRD
CrossRef - Hollmann C, Wiese T, Dennstädt F, Fink J, Schneider-Schaulies J, Beyersdorf N. Translational Approaches Targeting Ceramide Generation From Sphingomyelin in T Cells to Modulate Immunity in Humans. Front Immunol. 2019;10:475849. doi:10.3389/FIMMU.2019.02363/REFERENCE
CrossRef - Mori L, Lepore M, De Libero G. The Immunology of CD1- and MR1-Restricted T Cells. Annu Rev Immunol. 2016;34(Volume 34, 2016):479-510. doi:10.1146/ANNUREV-IMMUNOL-032414-112008/CITE/REFWORKS
CrossRef - Pereira CS, Macedo MF. CD1-Restricted T Cells at the Crossroad of Innate and Adaptive Immunity. J Immunol Res. 2016;2016(1):2876275. doi:10.1155/2016/2876275
CrossRef - Pereira CS, Sa-Miranda C, De Libero G, Mori L, Macedo MF. Globotriaosylceramide inhibits iNKT-cell activation in a CD1d-dependent manner. Eur J Immunol. 2016;46(1):147-153. doi:10.1002/EJI.201545725
CrossRef - Nieuwenhuis EES, Matsumoto T, Exley M, et al. CD1d-dependent macrophage-mediated clearance of Pseudomonas aeruginosa from lung. Nat Med. 2002;8(6):588-593. doi:10.1038/NM0602-588;KWRD
CrossRef - McGovern MM, Dionisi-Vici C, Giugliani R, et al. Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet Med. 2017;19(9):967-974. doi:10.1038/GIM.2017.7
CrossRef - Nascimbeni F, Dionisi Vici C, Vespasiani Gentilucci U, et al. AISF update on the diagnosis and management of adult-onset lysosomal storage diseases with hepatic involvement. Dig Liver Dis. 2020;52(4):359-367. doi:10.1016/J.DLD.2019.12.005
CrossRef - Cappellini MD, Motta I, Barbato A, et al. Similarities and differences between Gaucher disease and acid sphingomyelinase deficiency: An algorithm to support the diagnosis. Eur J Intern Med. 2023;108:81-84. doi:10.1016/J.EJIM.2022.11.028
CrossRef - Schuchman EH, Wasserstein MP. Types A and B Niemann-Pick disease. Best Pract Res Clin Endocrinol Metab. 2015;29(2):237-247. doi:10.1016/J.BEEM.2014.10.002
CrossRef - Kuchar L, Sikora J, Gulinello ME, et al. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal Biochem. 2017;525:73-77. doi:10.1016/J.AB.2017.02.019
CrossRef - Legnini E, Orsini JJ, Mühl A, Johnson B, Dajnoki A, Bodamer OA. Analysis of Acid Sphingomyelinase Activity in Dried Blood Spots Using Tandem Mass Spectrometry. Ann Lab Med. 2012;32(5):319-323. doi:10.3343/ALM.2012.32.5.319
CrossRef - Wasserstein MP, Desnick RJ, Schuchman EH, et al. The Natural History of Type B Niemann-Pick Disease: Results From a 10-Year Longitudinal Study. Pediatrics. 2004;114(6):e672-e677. doi:10.1542/PEDS.2004-0887
CrossRef - Geberhiwot T, Wasserstein M, Wanninayake S, et al. Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann–Pick disease types A, B and A/B). Orphanet J Rare Dis. 2023;18(1):1-28. doi:10.1186/S13023-023-02686-6/TABLES/5
CrossRef - Schuchman EH, Desnick RJ. The impact of sphingomyelin on the pathophysiology and treatment response to olipudase alfa in acid sphingomyelinase deficiency. Genet Med Open. 2024;2(1-2):101888. doi:10.1016/j.ymgme.2016.12.008
CrossRef - Diaz GA, Jones SA, Scarpa M, et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet Med. 2022;24(10):2209. doi:10.1016/J.GIM.2022.08.011
CrossRef - McGovern MM, Wasserstein MP, Kirmse B, et al. Novel first-dose adverse drug reactions during a phase I trial of olipudase alfa (recombinant human acid sphingomyelinase) in adults with Niemann–Pick disease type B (acid sphingomyelinase deficiency). Genet Med. 2016;18(1):34-40. doi:10.1038/GIM.2015.24
CrossRef - Wasserstein MP, Jones SA, Soran H, et al. Successful within-patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency. Mol Genet Metab. 2015;116(1-2):88-97. doi:10.1016/J.YMGME.2015.05.013
CrossRef - Wasserstein RL, Schirm AL, Lazar NA. Moving to a World Beyond “p < 0.05.” Am Stat. 2019;73(sup1):1-19. doi:10.1080/00031305.2019.1583913
CrossRef - Melmed GY, Botwin GJ, Sobhani K, et al. Antibody Responses After SARS-CoV-2 mRNA Vaccination in Adults With Inflammatory Bowel Disease. https://doi.org/107326/M21-2483. 2021;174(12):1768-1770. doi:10.7326/M21-2483
CrossRef - Schuchman EH, Desnick RJ. Types A and B Niemann-Pick disease. Mol Genet Metab. 2017;120(1-2):27-33. doi:10.1016/j.ymgme.2016.12.008
CrossRef - Lipiński P, Kuchar L, Zakharova EY, Baydakova G V., Ługowska A, Tylki-Szymańska A. Chronic visceral acid sphingomyelinase deficiency (Niemann-Pick disease type B) in 16 Polish patients: long-term follow-up. Orphanet J Rare Dis 2019 141. 2019;14(1):55-. doi:10.1186/s13023-019-1029-1
CrossRef - Thurberg BL, Wasserstein MP, Schiano T, et al. Liver and skin histopathology in adults with acid sphingomyelinase deficiency (niemann-pick disease type B). Am J Surg Pathol. 2012;36(8):1234-1246. doi:10.1097/PAS.0B013E31825793FF
CrossRef - McGovern MM, Lippa N, Bagiella E, Schuchman EH, Desnick RJ, Wasserstein MP. Morbidity and mortality in type B Niemann–Pick disease. Genet Med. 2013;15(8):618-623. doi:10.1038/GIM.2013.4
CrossRef - McGovern MM, Avetisyan R, Sanson BJ, Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis. 2017;12(1):1-13. doi:10.1186/S13023-017-0572-X/FIGURES/1
CrossRef - Hollak CEM, de Sonnaville ESV, Cassiman D, et al. Acid sphingomyelinase (Asm) deficiency patients in The Netherlands and Belgium: Disease spectrum and natural course in attenuated patients. Mol Genet Metab. 2012;107(3):526-533. doi:10.1016/J.YMGME.2012.06.015
CrossRef - Lipiński P, Kuchar L, Zakharova EY, Baydakova G V., Ługowska A, Tylki-Szymańska A. Chronic visceral acid sphingomyelinase deficiency (Niemann-Pick disease type B) in 16 Polish patients: Long-term follow-up. Orphanet J Rare Dis. 2019;14(1):1-9. doi:10.1186/S13023-019-1029-1/TABLES/1
CrossRef - Wasserstein M, Dionisi-Vici C, Giugliani R, et al. Recommendations for clinical monitoring of patients with acid sphingomyelinase deficiency (ASMD). Mol Genet Metab. 2019;126(2):98-105. doi:10.1016/J.YMGME.2018.11.014
CrossRef - Cox GF, Clarke LA, Giugliani R, McGovern MM. Burden of Illness in Acid Sphingomyelinase Deficiency: A Retrospective Chart Review of 100 Patients. JIMD Rep. 2018;41:119-129. doi:10.1007/8904_2018_120
CrossRef - McGovern MM, Wasserstein MP, Bembi B, et al. Prospective study of the natural history of chronic acid sphingomyelinase deficiency in children and adults: eleven years of observation. Orphanet J Rare Dis. 2021;16(1):1-14. doi:10.1186/S13023-021-01842-0/TABLES/5
CrossRef

