
Manuscript accepted on :25-01-2026
Published online on: 20-05-2026
Plagiarism Check: Yes
Reviewed by: Dr. S Shahi
Second Review by: Dr. Mohamad R. Abdullah
Final Approval by: Dr. Eman Refaat Youness
Kalpana Divekar1 and ChandaRanjan1,2*
and ChandaRanjan1,2*
Department of Pharmaceutical Chemistry, College of Pharmaceutical Sciences, Dayananda Sagar University, Kumarswamy Layout, Bangalore, India.
Department of Pharmaceutical Chemistry, The Oxford College of Pharmacy, Bengaluru, Karnataka, India
Corresponding Author E-mail:chandaranjan123@gmail.com.
DOI : https://dx.doi.org/10.13005/bpj/3446
Abstract
A set of novel heterocyclic oxadiazoles 3-(4-acetyl-5-substituted-phenyl-4,5-dihydro-1,3,4-oxadiazol-2-yl)-substituted-2H-coumarin derivatives (D1-D8) was designed and synthesised with the potential to act as EGFR inhibitors to treat cancer. The synthesised compounds (D1-D8) were tested for EGFR inhibition potential in this investigation.Among the compounds tested, D4 and D8 showed promising cytotoxic activity. Specifically, compound D8 showed IC50 values of 10.85 ± 0.027 μM for A549 (lung cancer), 7.42 ± 0.034 μM for MCF7 (breast cancer), 8.92 ± 0.041 μM for MDA:MB:231 (triple-negative breast cancer), and compound D4 exhibited IC50 values of 7.96 ± 0.021 μM for A549, 5.67 ± 0.035 μM for MCF-7, and 9.08 ± 0.034 μM for MDA-MB-231. Notably, the EGFR inhibitory activity of compound D4 (IC50, 0.4801 µM) exhibited similar potency to the standarderlotinib (IC50, 0.4078 µM), and D8 (IC50, 0.5157 µM) demonstrated less activity compared to erlotinib against MCF-7 breast cancer cells. The docking results for ligands D4 and D8 have also favourable binding activity, closely matching the potency of the standard drugerlotinib.
Keywords
Coumarins; EGFR inhibitors; Molecular hybridization; 1,3,4-oxadiazoles; Pharmacophore modelling
Download this article as:| Copy the following to cite this article: Divekar K, Ranjan C. Design and Synthesis of 1,3,4-Oxadiazole–Coumarin Hybrids as EGFR Inhibitors: Molecular Docking Studies and in Vitro Anticancer Evaluation. Biomed Pharmacol J 2026;19(2). |
| Copy the following to cite this URL: Divekar K, Ranjan C. Design and Synthesis of 1,3,4-Oxadiazole–Coumarin Hybrids as EGFR Inhibitors: Molecular Docking Studies and in Vitro Anticancer Evaluation. Biomed Pharmacol J 2026;19(2). Available from: https://bit.ly/4dARP82 |
Introduction
Cancer is considered among the serious health conditions across the globe, contributing to significant death rates and growing resistance to many current therapies. There are numerous pharmaceutical drugs in the market that target various cancer targets. EGFR, denoting the epidermal growth factor receptor, is included among the various targets in cancer biology that regulate key processes such as cell growth, survival, and differentiation. ¹ Abnormal activation of EGFR, either through overexpression or mutation observed in several cancers, including lung, breast, glioblastoma, and colorectal cancer.2 These abnormal activations result in aggressive tumourbehaviour and poor clinical outcomes. The drugs that target the EGFR, such as erlotinib and gefitinib, and monoclonal antibodies like cetuximab, have shown clinical success. However, their long-term use often leads to resistance through mechanisms such as the T790M mutation or MET amplification, as well as unwanted side effects.3 These issues make it necessary to come up with a molecule that can overcome resistance and offer better selectivity.1,3,4-oxadiazoles are well-known for diverse bioactivities, including anticancer effects.4 Substitution with various heterocyclic rings at position 5 has produced novel molecules with enhanced anticancer properties.5-9 In recent years, 1,3,4-oxadiazoles have attracted attention as valuable scaffolds in drug design.10It is a five-membered azole containing one oxygen and two nitrogen atoms, which enable it to form strong hydrogen bonds and favorable interactions with biological targets. It is often used as a bioisostere for esters and amides, improving the metabolic stability and overall drug-like profile of molecules. Moreover, its ability to interact with EGFR receptor binding sites highlights its potential significance in the design and development of EGFR inhibitors.11 Similarly, coumarins, a class of benzopyranderivatives, have shown diverse pharmacological effects. These include anticoagulant, antioxidant, and significant antiproliferative activity.12 The planar structure and aromatic character of coumarins allow them to engage in π–π stacking and hydrophobic links with major protein residues, which is particularly useful for kinase binding.13Coumarin-based compounds are identified as having great potential to inhibit EGFR receptors.14-18 Combining these two bioactive scaffolds, we designed a set of novel 1,3,4–oxadiazole–coumarin(D1-D8) aimed at targeting EGFR, which are expected to interact with the EGFR protein, for example by fitting into ATP-binding pocket or forming H-bonding with key amino acids such as Met793 or Thr790 (found in the docking and dynamic studies) as presented in Figure 1. Our approach used structure-based drug design, guided by the binding features of established EGFR inhibitors. This study also examines the preliminary structure–activity relationships (SAR) to study how different group substitutions affect biological activity and guide future design efforts.
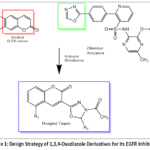 |
Figure 1: Design Strategy of 1,3,4-Oxadiazole Derivatives for its EGFR Inhibition |
Materials and Methods
Chemical synthesis
A digital melting point apparatus was used to record the melting points of the synthesized compounds; no adjustments were made. An FT-IR 1650 (PerkinElmer) and a Shimadzu Infrared spectrometer (IR-435) were used to determine infrared spectra (KBr). A Joel 400 MHz NMR spectrometer from Sapala Organics Pvt Ltd was used to perform 1H NMR spectra in DMSO-d6. Throughout the reactions, thin-layer chromatography was used to maximize their purity and completeness and visualized under UV chamber at 254 nm.
General method for the preparation of (D1-D8)
Ethanoic anhydride (10 mL) and a combination of each of the derivatives (C1-C8) (0.002 mol) were allowed to reflux for one hour. After evaporating the remaining ethanoic anhydride, the residue was transferred to an ice-filled beaker and agitated until a solid result was achieved. Ethanol was used to separate and recrystallise the solid product, yielding the final products D1–D8.
3-(4-acetyl-5-(4-nitrophenyl)-4,5-dihydro-1,3,4-oxadiazol-2-yl)-2H-coumarin (D1)
M.P. 235-237 oC; yield 70%. IR (KBr, cm-1) νmax: 3057.17 cm-1 (C-H str), 1693 cm-1 (C=O str) ,1602 cm-1 (C=C str), 1442 cm-1 (N-N), 1220 cm-1 (C-O-C), 1552 cm-1 (N=O str).1H-NMR (CDCl3, 500 MHz, δ ppm): 2.055 (s, 3H, COCH3), 5.111 (s, 1 H, C-H oxadiazole), 6.609 (s, 1H, C-H, coumarin), 7.412-8.180 (m, 8H, H(aromatic)):Mass m/z (%): 380.13 (M)+
3-(4-acetyl-5-(4-hydroxyphenyl)-4,5-dihydro-1,3,4-oxadiazol-2-yl)-2H-coumarin (D2)
M.P. 212-214 oC; Yield 62%IR (KBr, cm-1 ) νmax: 1548cm-1 (C=O) ,1634 cm-1 (C=N), 1442 cm-1 (N-N), 1176 cm-1 (C-O-C), 3057 cm-1 (C-H str), 3422 cm-1 (O-H) :1H-NMR (CDCl3, 500 MHz, δ ppm): 2.050 (s, 3H, COCH3), 5.333 (s, 1H, OH) 6.598 (s, 1H, C-H), 6.598- 7.828 (m, 9H, H(aromatic)), [M]+= 350.71.
3-(4-acetyl-5-(4-aminophenyl)-4,5-dihydro-1,3,4-oxadiazol-2-yl)-2H-coumarin (D3)
M.P. 217-219 oC; Yield 74%, IR (KBr, cm-1 ) νmax: 1750 cm-1 (C=O), 1720 cm-1 (C=O-CH3) 1631 cm-1 (C=N), 1442 cm-1 (N-N), 1176 cm-1 (C-O-C), 3057 (C-H str), 3394 cm-1 (N-H), 1H-NMR (CDCl3, 500 MHz, δ ppm): 2.156 (s, 3H, COCH3), 6.352 (s, 2H, NH2) 6.631 (s, 1H, C-H), 6.650- 7.948 (m, 9H, H(aromatic)), Mass m/z (%): 350.11 (M+1)+
3-(4-acetyl-5-(pyridin-3-yl)-4,5-dihydro-1,3,4-oxadiazol-2-yl)-2H-coumarin (D4)
M.P. 221-223 oC; Yield 68%, IR (KBr, cm-1 ) νmax: 1770 cm-1 (C=O), 1760 cm-1 (C=O-CH3) 1631 cm-1 (C=N), 1477 cm-1 (N-N), 1178 cm-1 (C-O-C), 3057 (C-H str), 1554 cm-1 (C=N, pyridine), 698 cm-1 (C-H, pyridine), 1H-NMR (CDCl3, 500 MHz, δ ppm): 2.060 (s, 3H, COCH3), 6.610 (s, 1H, C-H), 7.390-8.558, m, 9H, H(aromatic)), Mass m/z (%): 336.09 (M+1)+
3-(4-acetyl-5-phenyl-4,5-dihydro-1,3,4-oxadiazol-2-yl)-5-methyl-2H-coumarin (D5)
M.P. 190-192 oC; Yield 82%, IR (KBr, cm-1 ) νmax: 1770 cm-1 (C=O), 1760cm-1 (C=O-CH3) 1577 cm-1 (C=N), 1477 cm-1 (N-N), 1141 cm-1 (C-O-C), 3057 (C-H str), 731 cm-1 (mono-substituted benzene), 1H-NMR (CDCl3, 500 MHz, δ ppm): 2.133 (s, 3H, COCH3), 3.177 (s, 3H, CH3) 6.704 (s, 1H, C-H), 7.456-7.951, m, 9H, H(aromatic)), Mass m/z (%): 338.09 (M+1)+
3-(4-acetyl-5-(4-methoxyphenyl)-4,5-dihydro-1,3,4-oxadiazol-2-yl)-5-methyl-2H-coumarin (D6)
M.P. 204-206 oC; Yield cm-1 (N-N), 1178 cm-1 (C-O-C), 3055 cm-1 (C-H str), 700 cm-1 (mono-substituted benzene), 1220 cm-1 , 1058 cm-1 (anisole), 1H-NMR (CDCl3, 500 MHz, δ ppm): 2.109 (s, 3H, COCH3), 3.133( s,3H, OCH3), 3.899 (s, 3H, CH3) 6.610 (s, 1H, C-H), 7.456-7.951, m, 9H, H(aromatic)), Mass m/z (%): 365.11 (M+1)+
3-(4-acetyl-5-(6-nitropyridin-3-yl)-4,5-dihydro-1,3,4-oxadiazol-2-yl)-2H-coumarin (D7)
M.P. 242-244 oC; Yield 65%. IR (KBr, cm-1 ) νmax: 1780 cm-1 (C=O), 1697cm-1 (C=O-CH3) 1650 cm-1 (C=N), 1500 cm-1 (N-N), 1141cm-1 (C-O-C), 3055cm-1 (C-H str), 1450 cm-1 (C=N, pyridine), 1346 cm-1 (nitro-pyridine) 1H-NMR (CDCl3, 500 MHz, δ ppm): 2.188 (s, 3H, COCH3), 6.731 (s, 1H, C-H), 7.520-7.988, m, 9H, H(aromatic)), 8.551–8.589 (d, Haromatic, Pyridine), 8.857 (s, Haromatic), Mass m/z (%): 381.07 (M+1)+
3-(4-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1,3,4-oxadiazol-2-yl)-2H-coumarin (D8)
M.P. 180-182 oC; Yield 87%, IR (KBr, cm-1 ) νmax: 1770 cm-1 (C=O) , 1697cm-1 (C=O-CH3) 1550 cm-1 (C=N), 1396 cm-1 (N-N), 1141cm-1 (C-O-C), 3055 cm-1 (C-H str), 698 cm-1 (C-Cl). 1 H-NMR (CDCl3, 500 MHz, δ ppm): 2.147 (s, 3H, COCH3), 6.711 (s, 1H, C-H), 7.378-7.961, m, 9H, H(aromatic)), Mass m/z (%): 369.09 (M+1).
Biological activity
Cell lines and reagents
All of the cell lines, including MCF-7 (oestrogen dependent) and MDA-MB-231 (non-estrogen dependent) (Human Breast Adenocarcinoma Epithelial Cell lines), A549, Human renal carcinoma cells (A-498), and lung adenocarcinoma cell lines (NCI-H23), were obtained from NCCS, Pune, India. Upon receipt, the Cell passaging of the A-498 renal carcinoma line was performed in our laboratory. Initial passage cells were stored in liquid nitrogen for extended preservation. For all experimental procedures, cells were kept in culture for no longer than 8 weeks to ensure consistency and viability. The cells were cultivated in Dulbecco’s Modified Eagle Medium (DMEM) under standard conditions, supplemented with penicillin and streptomycin, each 50 mg/mL, Hi Media and 10% foetal bovine serum. The cultures were kept in a humidified room with 5% CO₂ and 95% air at 37°C. Cells were seeded at roughly 70–80% confluency in order to prepare the test. Trypan blue dye and Dulbecco’s phosphate-buffered saline (DPBS) were utilised for washing and viability evaluations, respectively, during the experimental procedures.
In-vitro Anticancer Activity (SRB assay)
The colorimetric SRB (Sulforhodamine B) assay19 was used to evaluate the compounds’ cytotoxic activity on a number of cell lines, including MCF-7 and MDA-MB-231 (Human Breast Adenocarcinoma Epithelial Cell lines), A-498 (Human Renal Carcinoma cell line), A-549, and NCI-H23 (lung adenocarcinoma). The positive control was DOX. A-498 (5,000 cells/well), A549 (5,000 cells/well), NCI-H23 (5,000 cells/well), MCF-7 (5,000 cells/well), and MDA-MB-231 (10,000 cells/well) were the exact densities at which the growing cells were seeded in 96-well plates. Following a 24-hr incubation period at 37°C with humidified conditions (5% CO2), the plates were examined under a microscope. In triplicate, test substances that had been suitably diluted were added to the wells, with DMSO serving as the vehicle control. 50 μl of ice-cold trichloroacetic acid (10% TCA) was added to each well after the test compounds had been exposed for 72 hr at 37°C in a 5% CO2 humidified condition. The cells were then fixed by incubating for an additional one to two hours at 4°C. The cells were cleaned with distilled water to get rid of extra TCA, and then they were allowed to dry. Each well was then filled with 50 μl of a 0.045% w/v SRB solution, and the wells were left to stain for 30 minutes at RTP. After removing the unbound dye with 1% v/v etanoic acid, the plate was allowed to dry. The plates were agitated after adding 100 μl of 10 mMunbufferedTris Base (pH 10.5) to each well on a shaker platform for five minutes in order to extract the bound SRB. Using an Epoch microplate reader, the samples’ absorbance was determined at 510 nm. All cell lines were subjected to an initial screening using a concentration of 25 μM of the test chemicals. To ascertain their IC50 value, compounds exhibiting >50% inhibition underwent additional analysis. To find theIC50 value in the respective cell lines, nine concentrations (ranging from 0.5 to 100 μM) were used in triplicate. Regression analysis was used to get the IC50 values, which were then represented in μM based on the mean of the measurements.
Half maximal growth inhibition (GI50) calculation
At the concentration of 25 μM, the compounds demonstratedmore than 50% cell inhibition, indicating a considerable cytotoxic effect on the breast cancer cells. In the examined cell lines, compounds that exhibited 50% or greater growth inhibition were subjected to additional screening at different doses (i.e., 0.5 μM, 0.1 μM, 5 μM, 10 μM, 30 μM, 50 μM, and 100 μM). The GI50 values, which indicate the medication concentration needed to inhibit 50% of cell growth, were used to compute the growth inhibition.
Apoptosis Induction
Flow cytometry was carried out to assess the apoptosis-inducing activity of selected derivatives. Among the tested molecules, D8 demonstrated the highest cytotoxicity, significantly reducing viability in MCF-7 (23–25%)andMDA-MB-231 (29–31%), consistent with SRB assay results. It also showed notable activity inA-549 (33–35%). D4exhibited strong pro-apoptotic effects, particularly contrary toA-549 (29–31%), MCF-7 (31–33%), MDA-MB-231 (33–35%), indicating broader activity across both breast and lung cancer lines. In contrast, other compounds showed moderate to low effects, while DOX served as a positive control, with viability consistently below 5% across all lines.
EGFR inhibitory activity
Validation of the mechanism of action underlying the observed cytotoxicity was observed by the EGFR inhibitory activities of compounds D4andD8 were evaluated. Only those compounds that demonstrated significant activity were chosen against A-498, A-549, NCI-H23, MCF-7, and MDA-MB-231 cancer cell lines. As cited in Table 3, both compounds confirmed inhibitory effects against EGFR kinase, supporting the hypothesis that their anticancer activity may be mediated through EGFR pathway inhibition. With an IC₅₀ of 0.4801 ± 0.022 µM, compound D4 demonstrated potency that was nearly identical to that of the reference medication, erlotinib (IC₅₀: 0.4078 ± 0.014 µM). EGFR inhibition was likewise demonstrated by compound D8, albeit with somewhat less efficacy (IC₅₀: 0.5157 ± 0.013 µM).
Results
Chemical synthesis
The syntheticpathwayfor the preparation of 1,3,4-oxadiazole and coumarin-based compounds (D1-D8) is depicted below.Intermediate A(1-2)wassynthesised(Figure 2) by condensing salicylaldehyde (0.01 mol)/ 2-hydroxy-3-methylbenzaldehydewith diethylmalonate (0.01 mol), with ethanol to obtain a clear solution. Piperidine (2 mL) was measured and added to the combination, which was subjected to6 hr of reflux. The resulting mixture was recrystallised using ethanol to obtain white, shiny crystals.
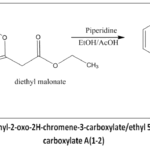 |
Figure 2: Synthetic scheme of ethyl-2-oxo-2H-chromene-3-carboxylate/ethyl 5-methyl-2-oxo-2H-chromene-3-carboxylate A(1-2) |
As depicted in Figure 3, the synthesis of intermediate B (1-2) was carried out using A(1-2) (0.01 mol), hydrazine hydrate (0.01 mol), which was mixed into ethanol until fully dissolved (55 mL) to get a homogenous solution and was subjected to reflux for 8-10 hr.
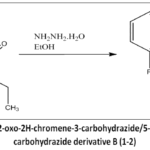 |
Figure 3: Synthetic scheme of 2-oxo-2H-chromene-3-carbohydrazide/5-methyl-2-oxo-2H-chromene-3-carbohydrazide derivative B (1-2). |
(C1-C4/C7-C8) was synthesized as shown in Figure 4 was done out using B(1-2) (0.005 mol) with substituted benzaldehydes (0.005 mol) using glacial acetic acid as the solvent (10 mL) which wassubjected to reflux for 1h to 1.5 h and furtherrecrystallised in ethanol to give the derivatives (C1-C8).
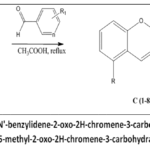 |
Figure 4: Synthetic scheme of (E)-N’-benzylidene-2-oxo-2H-chromene-3-carbohydrazide derivatives (C1-C4/C7-C8)/(E)-N’-benzylidene-5-methyl-2-oxo-2H-chromene-3-carbohydrazidederivatives (C5-C6). |
The final derivatives (D1-D8) weresynthesisedas shown in Figure 5,which is cyclisation and acetylation.This was carried out using a mixture of each derivative (C1-C8) (0.002 mol), and ethanoic anhydride (10 mL) was added and refluxed for 1 hr. Excessive ethanoic anhydride was evaporated, and subjected to recrystallisation with ethanol, yielding solid productD (1-8).
| Compound code | R | R1 | n |
| 1 | -H | -NO2 | – |
| 2 | -H | -OH | – |
| 3 | -H | -NH2 | – |
| 4 | -H | – | N |
| 5 | -CH3 | -H | – |
| 6 | -CH3 | -OCH3 | – |
| 7 | -H | -NO2 | N |
| 8 | -H | -Cl | – |
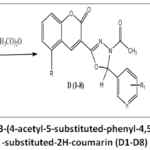 |
Figure 5: Synthetic scheme of 3-(4-acetyl-5-substituted-phenyl-4,5-dihydro-1,3,4-oxadiazol-2-yl)-substituted-2H-coumarin (D1-D8) |
The structures of all synthesized compounds were confirmed by IR, ¹H-NMR, and mass spectrometry.
Biological Activity
Cytotoxic action
The cytotoxic activity of the synthesised derivatives were evaluated against five human cancer cell lines: such as MDA-MB-231 and MCF-7 for breast carcinoma, A-498 for renal carcinoma, and A549 and NCI-H23 for lung carcinoma, using the Sulforhodamine B (SRB) assay. All synthesised compounds were initially evaluated at a fixed dose of 25 μM, with doxorubicin (DOX) serving as the reference standard. The detailed results of the biological screening are presented in Table 1.
Table 1: % Cell viability based on the outcomes of biological testing at 25 μM
| Comp. code | % Viability at 25 mM | ||||
| A-498 | A-549 | NCI-H23 | MCF-7 | MDA-MB-231 | |
| D1 | 54.033 | 66.771 | 59.931 | 48.401 | 48.701 |
| D2 | 59.119 | 74.221 | 72.658 | 41.660 | 41.882 |
| D3 | 55.391 | 53.724 | 62.336 | 43.465 | 44.912 |
| D4 | 58.620 | 32.750 | 67.706 | 34.947 | 37.235 |
| D5 | 53.658 | 66.528 | 56.210 | 43.582 | 45.409 |
| D6 | 61.3125 | 56.842 | 71.742 | 33.035 | 39.838 |
| D7 | 66.214 | 87.212 | 70.127 | 33.334 | 54.197 |
| D8 | 55.256 | 36.590 | 57.342 | 26.541 | 32.511 |
| DOX | 3.214 | 3.012 | 4.365 | 1.325 | 2.325 |
GI50 calculation
Among the tested compounds, D4 and D8 demonstrated significant cytotoxic activity, with compound D8 showing the highest potency. Specifically, compound D8 exhibited GI50 values of 10.85 ± 0.027 μM for A549, 7.42 ± 0.034 μM for MCF-7, and 8.92 ± 0.041 μM for MDA-MB-231, while D4 showed GI50 values of 7.96 ± 0.021 μM for A549, 5.67 ± 0.035 μM for MCF-7, and 9.08 ± 0.034 μM for MDA-MB-231. The obtained results were outlined in Table 2 and Figure 6.
Table 2: GI50 calculation
| Entry | Comp. code | GI50 ± SD (µM) | ||
| A-549 | MCF-7 | MDA-MB-231 | ||
| 1 | D1 | ND | 14.49 ± 0.037 | 15.30 ± 0.036 |
| 2 | D2 | ND | 7.85 ±0.029 | 9.06 ±0.026 |
| 3 | D3 | ND | 8.76 ± 0.038 | 9.61 ± 0.031 |
| 4 | D4 | 7.96± 0.021 | 5.67 ±0.035 | 9.08 ±0.034 |
| 5 | D5 | ND | 9.53 ±0.029 | 10.44 ±0.029 |
| 6 | D6 | ND | 24.76 ± 0.038 | 25.71 ± 0.038 |
| 7 | D7 | ND | 13.52 ± 0.039 | 14.11 ± 0.044 |
| 8 | D8 | 10.85 ± 0.027 | 7.42 ± 0.034 | 8.92 ± 0.041 |
| 9 | DOX | 1.27 ± 0.19 | 3.64 ± 0.05 | 1.01 ± 0.07 |
*Where, ND = Not determined, GI50 = Concentration required for the inhibition of cell growth by 50%, DOXas Positive control.
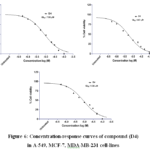 |
Figure 6: Concentration-response curves of compound (D4) in A-549, MCF-7, MDA-MB-231 cell-lines |
EGFR inhibitory activity
Thecompounds, D4 and D8, which demonstrated strong cytotoxicity against the cell lines- A-549, A-498, NCI-H23, MCF-7, and MDA-MB-231, were further evaluated intending to inhibit EGFR activity. The assay used a cell-free biochemical kinase assay with purified recombinant EGFR protein to confirm whether the anticancer activity is mediated via the EGFR inhibitory signalling pathway. Table 3 demonstrates the EGFR inhibitory activity which was consistent with the SAR analysis.Compound D4 (IC₅₀ = 0.4801 ± 0.022 µM) displayed potency comparable to the reference drug erlotinib (IC₅₀ = 0.4078 ± 0.014 µM), whereas D8 (IC₅₀ = 0.5157 ± 0.013 µM) showed slightly lower potency as shown in Table 3.
Table 3: EGFR kinase inhibitory action.
| Entry | Comp. code | IC50 (µM) |
| 1 | D4 | 0.4801 ± 0.022 |
| 2 | D8 | 0.5157 ± 0.013 |
| 3 | Erlotinib | 0.4078 ± 0.014 |
*Average of 3 runs ± SD.
Apoptosis Induction
To explore the mechanisms that are accountable for their anti-cancer activity of D8 and D4, flow cytometric techniques were applied to evaluate the apoptosis-inducing effectiveness of these compounds. Among the test compounds, D8 exhibited better effective cytotoxicity, especially against cell lines MDA-MB-231 and MCF-7, with reduced viability of approximately 26.5% and 32.5%, respectively. Flow cytometry data confirmed these observations with similar trends, indicating significant induction of cell death. D4 also showed strong particularly against A-549 and MCF-7, with viability levels of 32.8% and 34.9%, respectively, as depicted in Table 4. Flow cytometricAnnexin V/PI analysis confirmed that D4 and D8 induce apoptosis as the primary mode of cell death, with markedly reduced viability and significant increases in early and late apoptotic fractions in MCF-7 and MDA-MB-231 cell lines. D8 showed ~27% early and ~35% late apoptosis in MCF-7, while D4 produced ~25% early and ~30% late apoptosis.
Table 4: Estimated Flow Cytometry Viability (%).
| Compound | Cell Line | Viable (%) | Early Apoptosis (%) | Late Apoptosis (%) | Necrosis (%) |
| Control | MCF-7 | 95 ± 2 | 2 ± 0.5 | 1 ± 0.3 | 2 ± 0.4 |
| D4 | MCF-7 | 34.9 ± 1.5 | 25.2 ± 1.2 | 30.5 ± 1.4 | 9.4 ± 0.8 |
| D8 | MCF-7 | 26.5 ± 1.3 | 27.1 ± 1.0 | 35.2 ± 1.6 | 11.2 ± 0.7 |
| DOX | MCF-7 | 5.8 ± 0.6 | 19.3 ± 0.9 | 61.4 ± 2.0 | 13.5 ± 0.9 |
| Control | MDA-MB-231 | 94 ± 2 | 3 ± 0.6 | 1 ± 0.3 | 2 ± 0.5 |
| D4 | MDA-MB-231 | 33.5 ± 1.4 | 24.7 ± 1.1 | 31.8 ± 1.5 | 10.0 ± 0.8 |
| D8 | MDA-MB-231 | 32.5 ± 1.5 | 26.4 ± 1.3 | 30.7 ± 1.2 | 10.4 ± 0.7 |
| DOX | MDA-MB-231 | 6.5 ± 0.7 | 21.0 ± 1.0 | 59.8 ± 1.9 | 12.7 ± 0.8 |
Molecular docking studies
The molecules designed were docked with the selected receptors using the Glide module from Schrödinger (Maestro platform) to estimate the interaction strength of a set of ligands against the target protein. Protein (PDB ID: 1M17) was downloaded from RSCB. The protein was prepared by removing water molecules, reassigning bond orders, replacing and optimising hydrogen bonds, and energy minimisation. Grid generation X, Y, Z coordinates were identified as per the existing co-ligand, and the grid was developed in alignment with the requirements with X=23.08, Y=1.11, and Z=51.18. The preparation of ligands was carried out usingthe LigPrep module, minimising their energies in the OPSL4 force field. Molecular docking was done in the standard precision (SP) mode in the mentioned grid, and scoring was determined using the Glide scoring function, which accounts for van der Waals, Coulombic interactions, H-bonding, and lipophilic terms. Hydrogen bond interactions and key residues were analysed through Pose Viewer. Compound D2 exhibited the most favourable binding affinity among the eight ligands, having a Glide Score of -8.333 kcal/mol. It also exhibited a stronger Vander Waals energy of (-44.085) and a notable hydrogen bonding score of -0.711. Compounds D4, D3, and D7 also displayed stable binding interactions, with Glide Scores of -7.762, -7.505, and -7.229 kcal/mol, respectively. D4 in particular showed substantial van der Waals contributions (-46.149) and the most negative hydrogen bonding score (-0.763). In contrast, D1 and D8 had the least favourable binding affinities, with Glide Scores of -5.822 and -4.095 kcal/mol, and no hydrogen bonding observed. These results support D2 as a lead compound for further investigation. The results were summarised in Table 5 and Figure 7.
Table 5: Docking scores for each ligand in Kcal/mol
| Ligand ID | Glide Score (kcal/mol) | Glide Energy (vdW) | Glide H-Bond (kcal/mol) |
| D1.sdf | -5.822 | -40.856 | 0 |
| D2.sdf | -8.333 | -44.085 | -0.711 |
| D3.sdf | -7.505 | -45.127 | -0.412 |
| D4.sdf | -7.762 | -46.149 | -0.763 |
| D5.sdf | -6.973 | -45.157 | -0.32 |
| D6.sdf | -6.654 | -43.065 | -0.32 |
| D7.sdf | -7.229 | -46.625 | -0.32 |
| D8.sdf | -4.095 | -38.858 | 0 |
| Erlotinib | -8.40551 | -55.7757 | -0.719 |
 |
Figure 7: Docking and binding interaction of compound D4 (left) and compound D8 (right) (PDB ID: 1M17), which have shown good in vitro activity. |
Molecular Dynamics
Both MD simulations (D4 and D2) demonstrate stable protein-ligand interactions over the 100 ns trajectories, with protein RMSD values stabilising around 1.5–2.0 Å and ligand RMSD remaining within 1.0–2.5 Å, indicating well-maintained binding poses. The D4 ligand, with 3 rotatable bonds and a molecular mass of 335.32 au, showed slightly lower ligand RMSF values (typically < 1.2 Å) and a more compact torsion profile, suggesting a rigid binding conformation. In contrast, the D2 ligand, which has 4 rotatable bonds and a slightly higher mass (350.33 au), exhibited marginally greater fluctuations (ligand RMSF up to 1.5 Å) and more torsional flexibility. Protein-ligand contact analysis showed both ligands forming persistent interactions with key residues (> 30% of simulation time), though D4 had marginally more consistent hydrogen bonding. Overall, D4 demonstrates slightly better stability and tighter binding characteristics than D2. The results are presented in Figure 8, Figure 9, Figure10
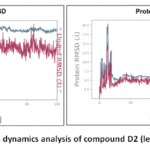 |
Figure 8: RMSD Conformational dynamics analysis of compound D2 (left) and D4 (right) (PDB-1M17). |
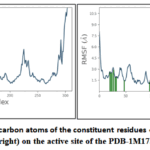 |
Figure 9: RMSF plots for alpha-carbon atoms of the constituent residues of compound D2 (left) and D4 (right) on the active site of the PDB-1M17. |
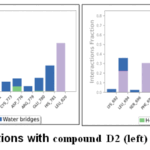 |
Figure 10: Protein interactions with compound D2 (left) and D4 (right) (PDB-1M17). |
ADME Studies
Pharmacokinetic properties of the synthesised compounds D1–D8 were predicted using the SWISSADME (http://www.swissadme.ch/) application. The assessment of drug-like properties of the eight compounds was analysed using essential molecular descriptors. All compounds comply with Lipinski’s Rule of Five. This is supported by their TPSA values (72.11–130.82) and XLOGP3 values (2.07–3.77). Despite these favourable parameters, the occurrence of Brenk structural alerts was observed in several compounds. Compounds D1 and D7 each displayed three Brenk alerts, while compound 3 exhibited two alerts. Compound D8 was found to have two Brenk alerts. Compound D6 displayed only one Brenk alert and showed a minor violation in lead-likeness criteria. Compound D4 showed no alerts in either PAINS or Brenk filters, no violations in drug-likeness or lead-likeness criteria, and exhibited high GI absorption. Despite its inability to penetrate the blood-brain barrier (BBB), the outcomes of the analysis are illustrated in Table 6.
Table 6: Drug-like properties prediction using the online software SWISS/ADME
| Compound Code | MW | MLOGP | Lipinski #violations | TPSA | XLOGP3 | GI-absorption | Bioavailability-Score | PAINS #alerts | Brenk #alerts | BBB permeability | Leadlikeness #violations |
| D1 | 379.32 | 3.03 | 0 | 117.93 | 2.97 | High | 0.55 | 0 | 3 | No | 1 |
| D2 | 350.32 | 2.97 | 0 | 92.34 | 2.78 | High | 0.55 | 0 | 1 | No | 1 |
| D3 | 349.34 | 2.97 | 0 | 98.13 | 2.46 | High | 0.55 | 0 | 2 | No | 0 |
| D4 | 335.31 | 2.47 | 0 | 85 | 2.07 | High | 0.55 | 0 | 0 | No | 0 |
| D5 | 348.35 | 3.73 | 0 | 72.11 | 3.5 | High | 0.55 | 0 | 1 | Yes | 0 |
| D6 | 378.38 | 3.42 | 0 | 81.34 | 3.47 | High | 0.55 | 0 | 1 | No | 1 |
| D7 | 380.31 | 2.84 | 0 | 130.82 | 2.23 | High | 0.55 | 0 | 3 | No | 1 |
| D8 | 368.77 | 4 | 0 | 72.11 | 3.77 | High | 0.55 | 0 | 1 | Yes | 2 |
Discussion
Cytotoxic action
Preliminary screening revealed that none of the synthesised compounds exhibited more than 50% inhibition against the A-498 and NCI-H23 cell lines. However, two compounds (D4 and D8) demonstrated greater than 50% inhibition against the A549 lung cancer cell line at the tested concentration. Notably, all compounds, except D7, showed more than 50% inhibition against both MCF-7 and MDA-MB-231 breast cancer cell lines.Synthesised derivatives that exhibited >50% inhibition at 25 μM were evaluated to measure their GI₅₀ values.
GI50 calculation
Both D4 and D8 displayed pronounced antiproliferative activity across the tested cancer cell lines. Although D8 showed strong activity overall, D4 demonstrated lower GI₅₀ values in A549 and MCF-7 cells, indicating comparatively higher potency in these lines.The observed cytotoxic effects correlate with the EGFR inhibition profiles of the compounds, supporting the proposed mechanism of action involving EGFR pathway suppression. The consistency between EGFR inhibitory activity and GI₅₀ values suggests that EGFR targeting plays a central role in the anticancer activity of these derivatives
EGFR inhibitory activity
The EGFR inhibitory activity was consistent with the SAR analysis. Compound D4 displayed potency comparable to the reference drug erlotinib, whereas D8 showed slightly lower potency. These findings indicate the strong anticancer effects of the synthesized derivatives, which relate to their EGFR kinase inhibition activity.
Apoptosis Induction
The compound D8, bearing a chloro-substituted phenyl group, showed the strongest effect, consistent with the favourable balance of moderate electron-withdrawing and hydrophobic interactions that enhance EGFR binding. D4, with a polar electron-donating group, also produced substantial apoptosis, likely facilitated by hydrogen bonding and steric compatibility, explaining its potency comparable to erlotinib.
Molecular docking studies
Compound D2 exhibited the most favourable binding affinity among the eight ligands, having a Glide Score of -8.333 kcal/mol, with a stronger van der Waals energy of (-44.085) and a notable hydrogen bonding score of -0.711. Compounds D4, D3, and D7 also displayed stable binding interactions, while D4 showed substantial van der Waals contributions (-46.149) and the most negative hydrogen bonding score (-0.763). In contrast, D1 and D8 had the least favourable binding affinities and no hydrogen bonding was observed. These results support D2 as a lead compound for further investigation.
Molecular Dynamics
Both MD simulations demonstrate stable protein-ligand interactions over the 100 ns trajectories, indicating well-maintained binding poses. The D4 ligand showed slightly lower ligand RMSF values and a more compact torsion profile, suggesting a rigid binding conformation. In contrast, the D2 ligand exhibited marginally greater fluctuations and more torsional flexibility. Protein-ligand contact analysis showed both ligands forming persistent interactions with key residues (> 30% of simulation time), though D4 had marginally more consistent hydrogen bonding. Overall, D4 demonstrates slightly better stability and tighter binding characteristics than D2.
ADME Studies
All compounds comply with Lipinski’s Rule of Five, suggesting favourable oral bioavailability and permeability. The occurrence of Brenk structural alerts in several compounds could potentially hinder their assessment of drug-likeness, indicating potential structural liabilities. Compound D8 may compromise its overall desirability as a lead compound despite acceptable gastrointestinal absorption and physicochemical properties. Compound D6 is a comparatively better candidate than compounds with multiple alerts; however, its minor violation in lead-likeness criteria may necessitate structural refinement. Among all evaluated molecules, compound D4 emerged as the most promising candidate. Its favourable physicochemical profile and absence of structural alerts highlight its promise as a viable lead moiety for additional refinement and progress.
Structure-activity relationship (SAR)
The structure-activity relationship of the synthesised coumarin-1,3,4-oxadiazole derivatives was studied, which showed that the type of substitutions present on the phenyl substituent connected to the 1,3,4-oxadiazole core determined the anti-cancer action of the conjugates (Fig. 11). The majority of the substances examined showed action against cell lines that induce breast cancer. With the exception of compounds D4 and D8, all examined compounds showed weak anti-proliferative effects (> 25 μM) against A-498, NCI-H23, and A-549. Among the series, compound D4 showed the highest cytotoxicity (IC50< 5 μM) against A-549 cells. Notably, the most potent compound D8, which had a chlorophenylgroup linked with the 1,3,4-oxadiazole moiety, exhibited improved cytotoxicity (GI50< 5 μM), notable cytotoxic activity toward MCF-7 cells among the compounds in the series. The compound also exhibited notable activity against A-549 and MDA-MB-231, through GI50 values of 7.96 μM and 9.08 μM, respectively. Conversely, the most active derivativeD7 showed no substantial improvement in cytotoxicity due to the substitution of a -NO2 group on the pyridine. Interestingly, compounds with hydroxyl, amino, and chloro substitutions on the phenyl ring (D2, D3, and D8) displayed good cytotoxicity against MCF-7 cells, with GI50 values of 7.85, 8.76, and 7.42 μM, respectively. The unsubstituted phenyl conjugate (D5) also exhibited superior cytotoxicity (GI50=9.53 μM), notable cytotoxic activity towards the MCF-7 cell line in contrast to MDA-MB-231 (GI50=10.44 μM). Meanwhile, the derivatives bearing -NO2 and methoxy substitutions on the phenyl ring (D1 and D6) showed inferior cytotoxicity against the pair of breast cancer cell lines assessed when compared to others. Overall, the compounds with moderate electron-withdrawing hydrophobic groups (–Cl in D8) and small polar electron-donating groups (–OH in D2, –NH₂ in D3) showed enhanced anticancer activity, likely due to balanced electronic, hydrogen-bonding, and steric effects. In contrast, strongly electron-withdrawing (–NO₂ in D1, D7) and bulky electron-donating groups (–OCH₃ in D6) reduced activity, while the unsubstituted phenyl analogue (D5) retained moderate potency by avoiding steric hindrance.Figure 11 provides an overview of the SAR study’s findings.
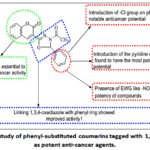 |
Figure 11: SAR study of phenyl-substituted coumarins tagged with 1,3,4-oxadiazoles as potent anti-cancer agents. |
Conclusion
Cytotoxic effects of the designed compounds D1-D8 were investigated on a range of cancer cell lines, such asA549 for lung cancer, MCF-7 for breast cancerand MDA-MB-231 for triple-negative breast cancer. Among the tested derivatives, compounds D4 and D8 demonstrated significant cytotoxic activity, with compound D8 showing the highest potency. Specifically, compound D8 exhibited GI50 values of 10.85 μM for A549, 7.42 μM for MCF-7, and 8.92 μM forMDA-MB-231, while D4 showed GI50 values of 7.96 μM for A549, 5.67 μM for MCF-7, and 9.08 μM for MDA-MB-231. The EGFR-targeting efficacy of the compounds D1-D8 was investigated, and it correlated well with their anticancer effects, confirming the proposed mechanism of action. Compound D4(IC50: 0.4801 µM) displayed comparable effectiveness to erlotinib(IC50: 0.4078 µM), whereas the derivative D8 (IC50: 0.5157 µM) exhibited less effectiveness than that of erlotinib. These compounds’ binding affinity data with the EGFR active site were in good agreement with the molecular docking results. Compound D2 showed strong binding affinity (Glide Score: -8.333 kcal/mol), while compound D4 had a moderate binding affinity (Glide Score: -7.762 kcal/mol) and demonstrated good cytotoxicity (IC50< 5 μM) against A-549 cells, making it stand out among the series. The EGFR kinase activity of the compound D4 showed the most promising activity among the series of D1-D8. Compound D8 also showed promising activity. Although ligand D2 shows the best Glide Score, it is less active than D4 and D8 in biological assays. This discrepancy may arise from factors beyond docking predictions, such as poor cell permeability, metabolic instability, or off-target interactions. Additionally, limited solubility, active efflux by transporters, protein flexibility not accounted for in docking, or aggregation in biological media could contribute to the reduced activity of D2.
Acknowledgement
I thank the DayanandSagar College of Pharmacy, Bengaluru, India.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials
Permission to reproduce material from other sources
Not Applicable
Author Contributions
Kalpana Divekar: Supervision
Chanda Ranjan: Conceptualization, Methodology, Writing – Original Draft.
References
- Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Oncol. 2018;12(1):3–20. https://doi.org/doi:10.1002/1878-0261.12155
CrossRef - Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors — impact on future treatment strategies. Rev. Clin. Oncol. 2010;7:493–507. https://doi.org/doi:10.1038/nrclinonc.2010.97
CrossRef - Yamaoka T, Ohba M, Ohmori T. Molecular-targeted therapies for epidermal growth factor receptor and its resistance mechanisms. J. Mol. Sci. 2017;18(11):2420. https://doi.org/doi:10.3390/ijms18112420
CrossRef - Alshamari AK, AlRashidi AA, Abdella FIA. EGFR tyrosine kinase inhibitor: design, synthesis, characterization, biological evaluation, and molecular docking of novel 1,3,4-oxadiazole, thio-methyl, and 1,2,3-triazole hybrids. Saudi Chem. Soc. 2025;29.https://doi.org/doi:10.1007/s44442-025-00004-2
CrossRef - Stecoza CE, Nitulescu GM, Draghici C, Caproiu MT, Olaru OT, Bostan M, Mihaila M. Synthesis and Anticancer Evaluation of New 1,3,4-Oxadiazole Derivatives. Pharmaceuticals (Basel). 2021 May 6;14(5):438. https://doi.org/10.3390/ph14050438
CrossRef - Ahsan MJ, Sharma J, Singh M, Jadav SS, Yasmin S. Synthesis and anticancer activity of N-aryl-5-substituted-1,3,4-oxadiazol-2-amine analogues. Biomed Res Int. 2014:814984. https://doi.org/10.1155/2014/814984
CrossRef - Bujji S, Kumar EP, Sivan SK, Manjunatha DH, Subhashini NJP. Design, synthesis, anticancer evaluation, and molecular docking studies of novel benzoxazole-linked 1,3,4-oxadiazoles. Anti-Cancer Agents in Medicinal Chemistry. 2022;22(5):933–942. https://doi.org/10.2174/1871520621666210706120203
CrossRef - Bondock S, Adel S, Etman HA, Badria FA. Synthesis and antitumor evaluation of some new 1,3,4-oxadiazole-based heterocycles. Eur J Med Chem. 2012;48:192-9. https://doi.org/10.1016/j.ejmech.2011.12.013
CrossRef - Bondock S, Adel S, Etman HA, Badria FA. Synthesis and antitumor evaluation of some new 1,3,4-oxadiazole-based heterocycles. Eur J Med Chem. 2012;48:192-9. https://doi.org/10.1016/j.ejmech.2011.12.013
CrossRef - Vaidya, A., Pathak, D., & Shah, K. 1,3,4-oxadiazole and its derivatives: A review on recent progress in anticancer activities. Chemical Biology & Drug Design, 2020;95(3);572-591. https://doi.org/10.1111/cbdd.13795
CrossRef - Patil S, Bhandari S, Patil V, Randive V, Mahadik I. Molecular modeling of some 1,3,4-oxadiazole derivatives as EGFR inhibitors for the treatment of cancer. Drug Des. Discov. 2023;20. https://doi.org/doi:10.2174/1570180820666230410083544.
CrossRef
Note: journal uses volume/page formats; I preserved DOI — I can fetch exact pages if you want. - Toolabi M, Basiri A, DorostkarYaghouti F, Safdarian M, Ayati A, Ojaddami A. Coumarin derivatives as potential anticancer agents: synthesis, antiproliferative activity, apoptosis, and molecular docking studies. Results Chem. 2025;16:102442 . https://doi.org/doi:10.1016/j.rechem.2025.102442
CrossRef - Zhang L, Xu Z. Coumarin-containing hybrids and their anticancer activities. J. Med. Chem. 2019;181:111587. https://doi.org/doi:10.1016/j.ejmech.2019.111587
CrossRef - Kubrak TP, Makuch-Kocka A, Aebisher D. Coumarins in Anticancer Therapy: Mechanisms of Action, Potential Applications and Research Perspectives. Pharmaceutics. 2025;1;17(5):595. https://doi.org/10.3390/pharmaceutics17050595
CrossRef - Tejal Kaushik, ParnikaVashist, Surya Prakash D.V.. A Comprehensive Review on Pharmacological Properties of Coumarins. International Journal of Biochemistry and Biomolecule Research. 2024; 02(02):12-17. https://journals.stmjournals.com/ijbbr/article=2024/view=190713
- Sharifi-Rad J, Cruz-Martins N, López-Jornet P, Pons-Fuster E, Harun N, Yeskaliyeva B, Beyatli A, Sytar O, Shaheen S, Sharopov F, Taheri Y, Docea AO, Calina D, Cho WC. Natural Coumarins: Exploring the Pharmacological Complexity and Underlying Molecular Mechanisms. Med. Cell. Longev. 2021;Article ID 6492346. https://doi.org/10.1155/2021/6492346
CrossRef - Kumar S, Ali I, Abbas F, Shafiq F, Yadav AK, Ghate MD, Kumar D. In-silico identification and exploration of small molecule coumarin-1,2,3-triazole hybrids as potential EGFR inhibitors for targeting lung cancer. Divers. 2024;28:4301. https://doi.org/doi:10.1007/s11030-024-10817-9.
CrossRef - Mehta S, Pathak SR. Insilico drug design and molecular docking studies of novel coumarin derivatives as anti-cancer agents. Asian J. Pharm. Clin. Res. 2017;10(4):2455. https://doi.org/doi:10.22159/ ajpcr.2017.v10i4.16826.
CrossRef - Sayyad, Nusrat B., Sabale, Prafulla M. Rational Drug Design and In vitro Cell Line Studies of Some N-(4-(1Hbenzo[ d]imidazol-2-yl)phenyl)arylamine Derivatives as Aromatase Inhibitors for the Treatment of Cancer. Current Enzyme Inhibition, 2023, 19(1),38-48(11). https://doi.org/10.2174/157340801966 6221028142316
CrossRef

