
Manuscript accepted on :16-03-2026
Published online on: 24-03-2026
Plagiarism Check: Yes
Reviewed by: Dr. Niharika Kondepudi and Dr. Raju Thenge
Second Review by: Dr. Akhundov Rashid Fatali
Final Approval by: Dr. Prabhishek Singh
Mehribon Pirimova1,2* , Masud Karimov2, Djalilov Abdulahat2, Feruz Ismoilov2, Muparrax Xodjayeva1, Oybek Sultonov2, Shokhida Keldiyorova3and Muhammad-Ali Mustafaev2
, Masud Karimov2, Djalilov Abdulahat2, Feruz Ismoilov2, Muparrax Xodjayeva1, Oybek Sultonov2, Shokhida Keldiyorova3and Muhammad-Ali Mustafaev2
1Department of Medical and Biological Chemistry, Tashkent State Medical University, Tashkent, Uzbekistan
2Tashkent Scientific Research Institute of Chemical Technology, Tashkent, Uzbekistan
3Department of Biological Chemistry, Samarkand State Medical University, Uzbekistan.
Corresponding Author E-mail: pirimova021293@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/3347
Abstract
Pyridine-2-carbothioamide and its Co(II), Ni(II), and Cu(II) complexes were evaluated as potential inhibitors of the ATPase domain of human topoisomerase IIα using an integrated density functional theory (DFT) and molecular docking approach. Geometry optimization and electronic-structure calculations showed that metal coordination markedly altered the frontier orbital distribution of the parent ligand, reduced the HOMO–LUMO energy gap, and increased global softness and electrophilicity. Docking simulations within the ATPase nucleotide-binding pocket indicated that all metal complexes displayed stronger predicted binding than the free ligand. Among the investigated systems, the Cu(II) complex exhibited the most favorable docking score and the richest interaction network with key active-site residues, including His120, Arg142, and Phe101. The observed trend suggests that coordination-induced electronic modulation contributes to enhanced binding capability within the catalytic pocket. Overall, the results identify pyridine-2-carbothioamide-based transition-metal complexes, particularly the Cu(II) derivative, as promising candidates for further experimental investigation as topoisomerase IIα-targeted metallodrugs. These findings provide a theoretical basis for the rational design of Topoisomerase IIα-targeted metallodrugs and highlight the importance of coordination-induced electronic modulation in enhancing ligand–protein interactions.
Keywords
Anticancer agents; Density functional theory; Molecular docking; Pyridine-2-carbothioamide complexes; Structure–activity relationship; Topoisomerase IIα.
Download this article as:| Copy the following to cite this article: Pirimova M, Karimov M, Abdulahat D, Ismoilov F, Xodjayeva M, Sultonov O, Keldiyorova S, Mustafaev M. A. Molecular Docking and DFT-Based Evaluation of Pyridine-2-Carbothioamide First-Row Transition-Metal Complexes Targeting Topoisomerase IIα. Biomed Pharmacol J 2026;19(1). |
| Copy the following to cite this URL: Pirimova M, Karimov M, Abdulahat D, Ismoilov F, Xodjayeva M, Sultonov O, Keldiyorova S, Mustafaev M. A. Molecular Docking and DFT-Based Evaluation of Pyridine-2-Carbothioamide First-Row Transition-Metal Complexes Targeting Topoisomerase IIα. Biomed Pharmacol J 2026;19(1). Available from: https://bit.ly/4tabsu1 |
Introduction
Background and significance
The development of biologically active metal complexes has emerged as an important direction in medicinal and bioinorganic chemistry owing to their tunable physicochemical properties and broad spectrum of biological activities.1 Coordination of organic ligands to transition-metal ions can significantly alter electronic structure, redox behavior, and molecular reactivity, often resulting in improved pharmacological properties compared with the corresponding free ligands.2 In particular, first-row transition metals such as copper(II), nickel(II), and cobalt(II) have attracted considerable interest due to their accessible oxidation states and flexible coordination geometries, which enable diverse modes of interaction with biological macromolecules.
Among the numerous ligand classes investigated for metallodrug development, sulfur-donor ligands have proven especially promising. Functional groups such as thioamides, thioureas, and dithiocarbamates display strong coordination ability toward transition metals and frequently exhibit enhanced biological activity upon metal binding.3,4 The soft and highly polarizable nature of sulfur donor atoms promotes favorable interactions with biological targets while simultaneously modifying the electronic environment of the metal center. Consequently, metal complexes containing sulfur-donor ligands have been widely reported to exhibit antimicrobial, antioxidant, and anticancer properties.3-5
Pyridine-2-carbothioamide (L) represents an attractive ligand scaffold because it contains a bidentate N,S-donor coordination system capable of forming stable complexes with transition metals. The ligand (C₆H₆N₂S) consists of a pyridine ring substituted with a carbothioamide functional group (–C(=S)NH₂), where the pyridine nitrogen and thioamide sulfur atoms act as potential donor sites. This arrangement enables efficient chelation to metal centers, typically forming five-membered metallacycles in transition-metal complexes. Such coordination frameworks can promote effective orbital overlap between the ligand and the metal center and thereby influence the electronic structure, reactivity, and biological behavior of the resulting complexes.
Previous studies on pyridine-derived thiosemicarbazone analogues have demonstrated that coordination with transition metals frequently enhances antiproliferative activity compared with the corresponding free ligands, with copper(II) complexes often exhibiting particularly strong cytotoxic effects.5,19 These findings highlight the important role of coordination-induced electronic modulation and intermolecular interactions in determining the biological activity of metal complexes.
Topoisomerase IIα as a biological target
DNA topoisomerase IIα (Topo IIα) is a clinically validated anticancer target that plays a central role in regulating DNA topology during replication and transcription.8 This homodimeric enzyme introduces transient double-strand breaks in DNA through an ATP-dependent catalytic cycle, allowing resolution of topological constraints generated during DNA metabolism.9 The N-terminal ATPase domain contains a conserved Bergerat fold, which forms the nucleotide-binding site responsible for ATP hydrolysis and enzymatic activation.10
Inhibition of the ATPase domain disrupts the catalytic cycle of Topo IIα and prevents DNA strand passage, ultimately leading to cytotoxic effects in rapidly proliferating cancer cells. Consequently, topoisomerase inhibitors represent an important class of anticancer drugs. In addition to classical organic inhibitors, metal-based complexes have recently attracted attention as potential Topo IIα inhibitors due to their ability to interact with both DNA and protein targets through multiple mechanisms, including ATPase inhibition, DNA intercalation, and redox-mediated damage.11
Despite the growing interest in metal-based Topo IIα inhibitors, the interaction of L-derived metal complexes with the ATPase catalytic pocket has not yet been systematically investigated. Understanding how coordination to transition metals influences electronic structure and protein binding behavior may therefore provide useful insights for the rational design of new metallodrug candidates targeting this enzyme.
Computational rationale and aim of the study
Computational chemistry methods provide powerful tools for investigating structure–activity relationships in metal-based drug candidates. Molecular docking enables prediction of ligand binding modes and interaction patterns within protein active sites, whereas density functional theory (DFT) provides detailed insight into electronic properties governing molecular reactivity and charge-transfer processes.12-15 Recent studies combining DFT calculations with docking simulations have demonstrated that coordination-induced changes in frontier molecular orbitals can influence the biological activity of metal complexes.16
According to frontier molecular orbital (FMO) theory, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) play key roles in determining electron-donating and electron-accepting capabilities of molecular systems.17,18 The HOMO–LUMO energy gap (ΔE) is frequently used as an indicator of molecular stability, polarizability, and charge-transfer potential, with smaller ΔE values generally corresponding to greater electronic flexibility.25 Experimental studies have demonstrated that copper(II) complexes of pyridine-derived thiosemicarbazones exhibit potent antiproliferative activity with IC50 values in the sub-micromolar range and high selectivity indices up to 5000.19 Similarly, nickel(II) thiosemicarbazone complexes have shown promising anticancer activity through both experimental and computational validation.20 Structural studies of thiosemicarbazone-based Ni(II), Cu(II), and related metal complexes have revealed correlations between coordination geometry and biological activity.21
Coordination of ligands to transition-metal centers often leads to redistribution of frontier molecular orbitals, resulting in reduced HOMO–LUMO gaps and increased electronic softness relative to the free ligand.22 Additionally, the redox activity of copper(II) complexes, particularly their ability to generate reactive oxygen species (ROS) through Cu(II)/Cu(I) cycling, represents an important additional mechanism for anticancer activity.34 In addition, molecular electrostatic potential (MEP) analysis can provide complementary information about electron density distribution and potential interaction sites within ligand structures.26
In the ATPase catalytic pocket of Topo IIα, residues such as His120, Arg142, Tyr105, and Phe101 can stabilize ligands through hydrogen bonding, electrostatic interactions, and aromatic stacking interactions.27 Consequently, coordination-induced changes in electronic structure may significantly influence the binding adaptability of metal complexes within this catalytic site.
Recent comprehensive reviews have highlighted the diverse biological applications of benzaldehyde-substituted thiosemicarbazones and their metal complexes, emphasizing structure–activity relationships and mechanistic insights.22 In this context, the present study investigates L and its Co(II), Ni(II), and Cu(II) complexes using an integrated computational approach that combines DFT calculations, molecular docking against the ATPase domain of human Topo IIα (PDB ID: 1ZXM), and MEP analysis. The aim of the present study is to investigate pyridine-2-carbothioamide and its Co(II), Ni(II), and Cu(II) complexes using an integrated computational framework combining density functional theory (DFT), molecular docking against the ATPase domain of human Topo IIα (PDB ID: 1ZXM), and molecular electrostatic potential (MEP) analysis. The study seeks to clarify how coordination-induced electronic modulation influences ligand–protein interactions within the ATPase nucleotide-binding pocket and to identify the most promising metal complex for further experimental investigation. To the best of our knowledge, this represents the first systematic computational analysis of pyridine-2-carbothioamide transition-metal complexes targeting the ATPase domain of human Topo IIα.
Materials and Methods
Computational chemistry
All quantum chemical calculations were carried out using Gaussian 16 (Revision C.01).28 The experimental crystal structure of L (CCDC 977685) was used as the starting geometry prior to full optimization.6 Geometry optimizations were performed at the B3LYP level of theory without symmetry constraints.29 Open-shell metal complexes were treated using the unrestricted UB3LYP formalism with appropriate spin states corresponding to the d⁷ Co(II), d⁸ Ni(II), and d⁹ Cu(II) electronic configurations. Frequency calculations were subsequently performed to confirm that all optimized structures corresponded to true minima on the potential energy surface, with no imaginary frequencies.
For non-metal atoms (C, H, N, and S), the 6-31G(d,p) basis set was employed, whereas the LANL2DZ effective core potential (ECP) was used for the metal centers (Co, Ni, and Cu).30,31 Solvent effects were incorporated using the polarizable continuum model (PCM) with DMSO as the dielectric medium in order to approximate a polar biological environment.32 Molecular orbital visualization was conducted using GaussView 6.33
Global reactivity descriptors, including chemical hardness (η), global softness (S), and electrophilicity index (ω), were derived from frontier orbital energies according to conceptual DFT equations.34 These descriptors were calculated using Koopmans’ approximation, where ionization potential and electron affinity were estimated from the HOMO and LUMO energies, respectively.
Protein and ligand preparation
The chemical structures of the ligand, reference nucleotides, and transition-metal complexes investigated in this study are presented in Figure 1. Table 1 summarizes the ligands, reference nucleotides, and transition-metal complexes considered in the present work together with their abbreviations, roles in the study, and molecular formulas.
Table 1: Ligands, reference nucleotides, and transition-metal complexes investigated in this study, with their abbreviations, roles, molecular/empirical formulae, and structural descriptions.
| Entry | Ligand / compound | Abbreviation used in text | Role in study | Molecular / empirical formula | Structural description |
| 1 | Pyridine-2-carbothioamide | L | Main ligand | C₆H₆N₂S | Pyridine ring substituted with a carbothioamide group; N,S-donor bidentate ligand |
| 2 | Phosphoaminophosphonic acid-adenylate ester | ANP | Co-crystallized reference ligand in PDB 1ZXM | C₁₀H₁₇N₆O₁₂P₃ | Crystallographic form of the non-hydrolysable ATP analogue bound in the Topo IIα ATPase site |
| 3 | Adenylyl-imidodiphosphate / AMP-PNP / AMPPNP | AMP-PNP | Common biochemical name of ANP | C₁₀H₁₇N₆O₁₂P₃* | Non-hydrolysable ATP analogue in which the bridging oxygen between the β- and γ-phosphates is replaced by an imido (–NH–) group |
| 4 | Adenosine triphosphate | ATP | Natural ligand used for docking control | C₁₀H₁₆N₅O₁₃P₃ | Endogenous substrate of the ATPase catalytic pocket |
| 5 | Dichlorobis(pyridine-2-carbothioamide)cobalt(II) | [CoL₂Cl₂] | Investigated metal complex | C₁₂H₁₂N₄S₂Cl₂Co | Co(II) complex with two N,S-chelating ligands |
| 6 | Dichlorobis(pyridine-2-carbothioamide)nickel(II) | [NiL₂Cl₂] | Investigated metal complex | C₁₂H₁₂N₄S₂Cl₂Ni | Ni(II) complex with two N,S-chelating ligands |
| 7 | Dichlorobis(pyridine-2-carbothioamide)copper(II) | [CuL₂Cl₂] | Investigated metal complex | C₁₂H₁₂N₄S₂Cl₂Cu | Cu(II) complex with two N,S-chelating ligands |
*For the crystallographic ligand deposited in the PDB, the chemical component is annotated as ANP and described as phosphoaminophosphonic acid-adenylate ester; AMP-PNP/AMPPNP is the commonly used biochemical name for the same non-hydrolysable ATP analogue.
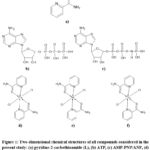 |
Figure 1: Two-dimensional chemical structures of all compounds considered in the present study: (a) pyridine-2-carbothioamide (L), (b) ATP, (c) AMP-PNP/ANP, (d) [CoL₂Cl₂], (e) [NiL₂Cl₂], and (f) [CuL₂Cl₂]. |
The crystal structure of the human Topo IIα ATPase domain was retrieved from the Protein Data Bank as PDB entry 1ZXM (resolution 1.87 Å).35 The structure corresponds to the N-terminal ATPase domain (residues 1–265) and contains a co-crystallized nucleotide analogue annotated in the PDB chemical component dictionary as ANP. In PDB nomenclature, ANP denotes phosphoaminophosphonic acid-adenylate ester with the empirical formula C₁₀H₁₇N₆O₁₂P₃; this ligand is the crystallographic representation of the widely used non-hydrolysable ATP analogue AMP-PNP (adenylyl-imidodiphosphate; AMPPNP). Thus, in the present manuscript, the terms ANP and AMP-PNP refer to the same nucleotide analogue, with ANP used when referring specifically to the ligand deposited in the 1ZXM crystal structure and AMP-PNP used as its common biochemical name.
Protein preparation was performed using AutoDockTools 4.36 The co-crystallized ANP ligand was removed for redocking validation, crystallographic water molecules were deleted except for structurally conserved ones, polar hydrogens were added, Kollman charges were assigned, and the receptor was converted to PDBQT format. The docking site was defined around the nucleotide-binding pocket occupied by ANP in the crystal structure.
The ligand set investigated in this study comprised L, the co-crystallized reference nucleotide analogue ANP/AMP-PNP, the natural substrate ATP, and the metal complexes [CoL₂Cl₂], [NiL₂Cl₂], and [CuL₂Cl₂]. The optimized geometries of L and the metal complexes obtained from DFT calculations were used for docking preparation. Ligands were processed in AutoDockTools 4 by adding Gasteiger charges, merging nonpolar hydrogens, and converting the structures to PDBQT format while retaining the DFT-optimized coordination geometries for the metal complexes.
Docking protocol, validation, and limitations
Molecular docking simulations were performed using AutoDock Vina v1.2.0.37,42 The grid box was defined to fully encompass the nucleotide-binding pocket and surrounding residues involved in ATP recognition in the crystallographic structure. The docking grid was centered on the nucleotide-binding pocket defined by the co-crystallized ANP ligand, and the grid box parameters used in the simulations are summarized in Table 2.
Table 2: Grid box parameters used for molecular docking simulations with AutoDock Vina in the ATPase domain of human Topo IIα (PDB ID: 1ZXM).
| Parameter | Value |
| Center x (Å) | 24.5 |
| Center y (Å) | −18.2 |
| Center z (Å) | 32.1 |
| Size x (Å) | 24 |
| Size y (Å) | 24 |
| Size z (Å) | 24 |
| Exhaustiveness | 8 |
| Number of poses per ligand | 20 |
| Energy range (kcal·mol⁻¹) | 3 |
The same grid box was applied to all ligands to ensure direct comparability of docking results. Docking calculations were carried out using the standard AutoDock Vina scoring function with an exhaustiveness value of 8, 20 poses generated per ligand, and an energy range of 3 kcal·mol⁻¹. The top-ranked pose with the lowest predicted binding energy was selected for further analysis, provided that it also exhibited chemically reasonable hydrogen-bonding and π–π interaction patterns within the ATPase pocket. In the docking simulations, metal centers were treated using standard AutoDock Vina atom parameters without explicit metal coordination constraints. Accordingly, docking scores were interpreted as relative comparative indicators rather than absolute thermodynamic binding free energies, because classical docking scoring functions do not explicitly account for several metal-specific effects, including coordination geometry preference, polarization, charge transfer, and redox behavior.38 Therefore, the docking analysis was used primarily to identify binding trends, pose geometries, and interaction patterns rather than to estimate exact binding energetics. The docking protocol was validated by redocking the co-crystallized ligand ANP, which corresponds to the non-hydrolysable ATP analogue AMP-PNP. The reproduced pose yielded a heavy-atom root-mean-square deviation (RMSD) of 1.74 Å, which is within the commonly accepted validation threshold of ≤2.0 Å, thereby supporting the reliability of the docking setup. As an additional biological relevance control, the natural substrate ATP was docked into the same pocket using identical parameters. ATP adopted a binding orientation overlapping with the crystallographic ANP pose and formed hydrogen bonds with key residues including His120, Asn163, and Lys168, supporting the relevance of the selected binding site for comparative docking analysis. The top-ranked docking pose (mode 1) was selected for each compound on the basis of the lowest predicted binding energy together with chemically reasonable interaction geometry within the nucleotide-binding pocket. Because the present study focuses on comparative analysis of the best-scoring binding modes, only the selected top-ranked poses are reported and discussed in the manuscript.
Interaction analysis
Ligand–protein interactions were analyzed using BIOVIA Discovery Studio Visualizer and PyMOL.39,40 Hydrogen bonds were identified using a donor–acceptor distance cutoff of ≤3.5 Å and a bond angle cutoff of ≥120°. π–π stacking interactions were evaluated on the basis of centroid-to-centroid distances of 3.5–5.0 Å. Interaction analysis was performed for the selected top-ranked docking pose of each ligand in order to compare residue-level binding patterns across the investigated systems.
MEP analysis
MEP surfaces were calculated at the same level of theory used for geometry optimization, namely B3LYP/6-31G(d,p)/LANL2DZ with PCM (DMSO). The electrostatic potential was mapped onto the electron density isosurface of 0.001 a.u., which approximates the molecular van der Waals surface. For consistent comparison among all structures, the potential scale was fixed between −0.05 and +0.05 a.u.. MEP analysis was used to visualize electron-density redistribution upon metal coordination and to identify regions likely to participate in electrostatic and hydrogen-bonding interactions with residues in the Topo IIα ATPase pocket.
Statistical analysis
The relationship between electronic descriptors and docking scores was evaluated using the Pearson correlation coefficient. Because the analysis involved only four systems (free ligand, Co(II), Ni(II), and Cu(II) complexes), the resulting correlations were interpreted cautiously as indicative trends rather than statistically definitive relationships.
Results and Discussion
Structural and electronic properties
Geometry optimization confirmed stable N,S-bidentate coordination of L with Co(II), Ni(II), and Cu(II) ions, producing well-defined chelate structures with bite angles of approximately 78–82°. Coordination occurs through the pyridine nitrogen and thioamide sulfur atoms, forming five-membered chelate rings that stabilize the metal center and enhance metal–ligand orbital overlap. This coordination mode promotes partial π-electron delocalization across the ligand backbone and the coordination framework, consistent with previous reports for sulfur-donor metal complexes.41
FMO analysis shows that metal coordination significantly modifies the electronic structure of the ligand. In particular, the HOMO–LUMO energy gap decreases progressively along the series:
Ligand (5.41 eV) > Co(II) (4.18 eV) > Ni(II) (3.92 eV) > Cu(II) (3.75 eV).
The frontier molecular orbital distributions and the schematic HOMO–LUMO energy level pattern for the investigated systems are illustrated in Figure 2.
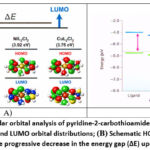 |
Figure 2: Frontier molecular orbital analysis of pyridine-2-carbothioamide (L) and its transition-metal complexes. (A) HOMO and LUMO orbital distributions; (B) Schematic HOMO–LUMO energy level diagram illustrating the progressive decrease in the energy gap (ΔE) upon metal coordination. |
The HOMO orbitals are mainly localized over the sulfur donor atoms and the conjugated pyridine ring, whereas the LUMO orbitals extend toward the metal center and the coordination framework, indicating possible charge-transfer interactions that may contribute to enhanced binding capability.
This systematic narrowing of ΔE correlates with increased electronic softness and improved predicted binding affinity observed in docking simulations. The calculated orbital energies and global reactivity descriptors are summarized in Table 3.
Table 3: Frontier molecular orbital energies and conceptual DFT-based global reactivity descriptors of L and its first-row transition-metal complexes.
| Compound | E_HOMO (eV) | E_LUMO (eV) | ΔE (eV) | Hardness η (eV) | Softness S (eV⁻¹) | Electrophilicity ω (eV) |
| Ligand (L) | −6.21 | −0.80 | 5.41 | 2.71 | 0.37 | 1.52 |
| [CoL₂Cl₂] | −5.68 | −1.50 | 4.18 | 2.09 | 0.48 | 2.11 |
| [NiL₂Cl₂] | −5.44 | −1.52 | 3.92 | 1.96 | 0.51 | 2.35 |
| [CuL₂Cl₂] | −5.31 | −1.56 | 3.75 | 1.88 | 0.53 | 2.61 |
Note: η = (E_LUMO − E_HOMO)/2, S = 1/η, and ω = μ²/(2η), where μ = (E_HOMO + E_LUMO)/2. Orbital energies and conceptual DFT descriptors were calculated at the B3LYP/6-31G(d,p)/LANL2DZ level with PCM (DMSO).
Docking validation and binding interactions
The reliability of the docking protocol was evaluated through re-docking of the co-crystallized nucleotide analog ANP into the ATPase binding pocket of Topo IIα. The reproduced pose showed an RMSD of 1.74 Å, which falls within the generally accepted validation threshold of ≤2.0 Å, confirming the accuracy of the docking procedure.
Docking simulations indicate that metal coordination significantly improves the predicted binding affinity of L toward the ATPase catalytic pocket. The predicted affinity trend follows:
Cu(II) > Ni(II) ≈ Co(II) > Ligand.
More negative docking scores correspond to stronger predicted binding affinity within the ATPase catalytic pocket. The free ligand exhibits the weakest predicted binding affinity (−5.2 kcal·mol⁻¹) and forms only a single hydrogen bond with Tyr105 together with hydrophobic contacts. In contrast, the metal complexes establish multiple stabilizing interactions with residues lining the nucleotide-binding pocket. The predicted binding orientations and interaction patterns of the ligand, its transition-metal complexes, and reference nucleotides within the ATPase domain of Topo IIα are illustrated in Figure 3, while the corresponding docking energies and interaction statistics are summarized in Table 4.
 |
Figure 3: Molecular docking analysis of pyridine-2-carbothioamide (L), its transition-metal complexes, and reference nucleotides within the ATPase domain of human Topo IIα (PDB ID: 1ZXM). |
Table 4: Molecular docking results and interaction analysis of L, its transition-metal complexes, ATP, and the co-crystallized reference ligand ANP within the ATPase domain of human Topo IIα (PDB ID: 1ZXM).
| Compound | Role in study | Selected pose (mode) | Best docking score (kcal·mol⁻¹) | RMSD l.b. (Å) | RMSD u.b. (Å) | H-bonds (n) | π–π interactions (n) | Hydrophobic contacts (n) | Key interacting residues |
| ANP | Redocking reference (co-crystallized ligand) | 1 | -9.1 | 1.74 | 1.98 | 3 | 1 | 2 | His120, Arg142, Tyr105 |
| ATP | Biological control (natural substrate) | 1 | -8.1 | – | – | 3 | 0 | 2 | His120, Asn163, Lys168 |
| L | Main ligand | 1 | -5.2 | – | – | 1 | 0 | 1 | Tyr105, Gly143 |
| [CoL₂Cl₂] | Investigated metal complex | 1 | -7.7 | – | – | 2 | 1 | 2 | His120, Asp176, Tyr105 |
| [NiL₂Cl₂] | Investigated metal complex | 1 | -7.7 | – | – | 3 | 1 | 2 | Lys89, His120, Phe101 |
| [CuL₂Cl₂] | Investigated metal complex | 1 | -7.8 | – | – | 4 | 2 | 3 | His120, Arg142, Phe101 |
Note: Docking calculations were performed using AutoDock Vina v1.2.0 with identical grid parameters for all compounds. The selected pose corresponds to the top-ranked mode used for interaction analysis in the main text. For the redocked co-crystallized ligand ANP, AutoDock Vina reports both RMSD lower bound (l.b.) and RMSD upper bound (u.b.) values relative to the crystallographic pose. RMSD values are not reported for ATP, L, and the metal complexes in the present table because these compounds were analyzed by comparative docking rather than by crystallographic pose reproduction. Hydrogen bonds were identified using donor–acceptor distances ≤3.5 Å and angles ≥120°, while π–π interactions were assigned using centroid–centroid distances of 3.5–5.0 Å. Only the selected top-ranked poses are reported because the present study focuses on comparative interpretation of the best-scoring binding modes.
Among the investigated systems, the Cu(II) complex exhibits the most favorable docking score (−7.8 kcal·mol⁻¹) and forms the most extensive interaction network. The complex forms several stabilizing contacts, including an S···H–N interaction with His120 (2.4 Å), hydrogen bonding between the pyridine nitrogen and Arg142 (2.8 Å), and additional contacts involving axial chloride ligands with residues such as Lys89 (3.1 Å) and Gly143 (3.3 Å). Furthermore, π–π stacking interactions occur between the pyridine ring and aromatic residues Phe101 (4.2 Å) and Tyr105 (4.5 Å).
The Ni(II) and Co(II) complexes display similar binding orientations but generally form slightly fewer stabilizing interactions and exhibit somewhat longer contact distances. These results suggest that the presence of a coordinated metal center enhances ligand–protein recognition by improving spatial orientation and enabling additional electrostatic and aromatic interactions within the ATPase catalytic pocket.
Electronic structure–binding relationship
A positive linear relationship was observed between the HOMO–LUMO energy gap (ΔE) and the predicted docking score. Compounds with smaller ΔE values exhibit more negative docking energies, indicating stronger predicted binding within the ATPase pocket. Linear regression analysis yielded the relationship y = 1.65x − 14.22 (R² = 0.96). This trend suggests that coordination-induced narrowing of the HOMO–LUMO energy gap may enhance the predicted binding affinity of the metal complexes.
The relationship between the HOMO–LUMO energy gap (ΔE) and the predicted docking affinity is illustrated in Figure 4, showing a clear linear trend between electronic structure and binding behavior.
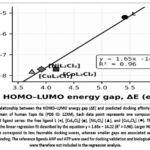 |
Figure 4: Relationship between the HOMO–LUMO energy gap (ΔE) and predicted docking affinity toward the ATPase domain of human Topo IIα (PDB ID: 1ZXM). Each data point represents one compound from the investigated ligand series: the free ligand L (●), [CoL₂Cl₂] (■), [NiL₂Cl₂] (▲), and [CuL₂Cl₂] (◆). |
From the perspective of conceptual DFT, the reduction in ΔE upon metal coordination corresponds to increased electronic softness and enhanced polarizability. These properties facilitate charge redistribution during intermolecular interactions, which may strengthen ligand–protein binding within the ATPase pocket.
Additional insight into interaction behavior was obtained through MEP analysis. In the free ligand, regions of negative electrostatic potential are concentrated mainly on the thioamide sulfur and pyridine nitrogen atoms, which act as potential hydrogen-bond acceptor sites. Upon coordination, electron density becomes more delocalized across the metal–ligand framework, particularly in the Cu(II) complex, which exhibits the most extended electron-rich surface. This redistribution is consistent with the observed decrease in ΔE and increase in electrophilicity. Given the limited number of investigated systems, the correlation should be regarded as a qualitative trend rather than a statistically robust relationship.
Taken together, the combined DFT, docking, and MEP results suggest a coordination-driven structure–activity relationship in which metal binding reduces the HOMO–LUMO gap, increases electronic softness, and enhances interaction capability within the Topo IIα ATPase catalytic pocket. Among the investigated systems, the Cu(II) complex shows the most pronounced electronic modulation and interaction network, consistent with its superior predicted docking affinity.
Strengths of the study
Strengths of the present study include the integrated analysis of electronic structure and ligand–protein interactions within a single computational framework. By combining density functional theory calculations, molecular docking, and electrostatic potential analysis, the study provides a mechanistic interpretation of how coordination-induced electronic modulation influences binding behavior in the ATPase pocket of Topo IIα.
In addition, the docking protocol was validated using both the co-crystallized nucleotide analogue ANP and the natural substrate ATP, which improves the reliability and biological relevance of the predicted binding trends for the investigated metal complexes.
Study limitations
Several methodological limitations should be considered when interpreting the present results. First, the study is entirely computational and predictive, and the docking-derived binding affinities represent relative trends that require experimental validation. Classical docking scoring functions have well-known limitations in describing transition-metal systems, particularly regarding metal coordination geometry, polarization effects, charge transfer, and redox chemistry.
Furthermore, solvation effects are treated differently in the DFT calculations (PCM-DMSO) and docking simulations (implicit solvent model), which may influence estimated interaction energies. The possible Cu(II)/Cu(I) redox cycling relevant to copper-based anticancer mechanisms was not explicitly modeled. In addition, the static docking approach does not account for protein flexibility or induced-fit effects within the ATPase binding pocket. The Cu(II)/Cu(I) redox couple can generate reactive oxygen species (ROS) through Fenton-type chemistry, contributing to oxidative stress-mediated cytotoxicity.22,34 This redox cycling may provide an additional anticancer mechanism beyond ATPase inhibition, though the relative contributions of these pathways require experimental investigation.19,20
Finally, the correlation analysis between electronic descriptors and docking affinity is based on a limited number of investigated systems (n = 4) and should therefore be interpreted as indicative rather than statistically definitive. Future studies should include experimental validation through enzyme inhibition assays, cytotoxicity testing against cancer cell lines, and DNA-binding investigations to confirm the predicted biological activity of these complexes. Another important limitation concerns the treatment of transition-metal centers in classical docking algorithms. AutoDock Vina employs simplified atom-type parameters and does not explicitly account for metal–ligand coordination geometry, polarization effects, or charge-transfer interactions that may occur in transition-metal complexes. Consequently, the predicted docking scores should be interpreted primarily as relative indicators of binding affinity rather than precise thermodynamic binding free energies.
Conclusion
This study presents a computational evaluation of pyridine-2-carbothioamide and its Co(II), Ni(II), and Cu(II) complexes using DFT, molecular docking, and MEP analysis. The results show that metal coordination significantly modifies the electronic structure of the ligand and improves its predicted interaction with the ATPase domain of human Topo IIα. Among the investigated systems, the Cu(II) complex exhibited the most favorable combination of reduced HOMO–LUMO gap, increased electrophilicity, and stronger docking interactions with key catalytic residues, including His120, Arg142, and Phe101. Although experimental validation is still required, the present results provide a useful theoretical framework for the design of Topo IIα-targeted metal-based inhibitors. Future studies will focus on enzymatic inhibition assays and cytotoxicity evaluation against relevant cancer cell lines. The present computational findings provide mechanistic insight into the structure–activity relationship of pyridine-2-carbothioamide metal complexes and support further experimental investigation of these systems as potential Topo IIα inhibitors.
Acknowledgement
The authors express their sincere gratitude to the Tashkent Chemical and Technological Scientific Research Institute, Uzbekistan, for providing laboratory infrastructure, technical facilities, and scientific support essential for conducting this research. The authors also acknowledge Tashkent State Medical University, Uzbekistan, for its academic environment and institutional support during the preparation of this manuscript. The authors greatly appreciate the professional guidance and research conditions that contributed to the successful completion of this study.
Funding sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed Biomedical and Pharmacology Journal consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials
Permission to reproduce material from other sources.
Not Applicable
Author Contributions
- Mehribon Pirimova: Conceptualization, Methodology, Investigation, Data curation, Writing – original draft, Writing – review & editing.
- Masud Karimov: Formal analysis, Software, Validation, Data curation.
- Djalilov Abdulahat: Supervision, Project administration, Writing – review & editing.
- Feruz Ismoilov: Methodology, Investigation, Visualization.
- Muparrax Xodjayeva: Investigation, Data curation, Writing – original draft.
- Oybek Sultonov: Methodology, Investigation, Visualization.
- Shokhida Keldiyorova: Resources, Data curation, Validation.
- Muhammad-Ali Mustafaev: Resources, Validation, Data curation.
References
- Yusof ENM, Ahmad R, Khan SU, et al. Structural and computational investigation of metal complexes. Journal of Molecular Structure. 2025;1440:144044. https://doi.org/10.1016/j.molstruc.2025.144044
CrossRef - Sharma N, Dhingra N, Singh HL. Design, spectral, antibacterial and in-silico studies of new thiosemicarbazones and semicarbazones derived from symmetrical chalcones. Journal of Molecular Structure. 2024;1307:138000. https://doi.org/10.1016/j.molstruc.2024.138000.
CrossRef - Oladipo SD, Muthuraj V, Govender T, et al. Insight into molecular interactions of bioactive compounds. Scientific Reports. 2023;13:39502. https://doi.org/10.1038/s41598-023-39502-x
CrossRef - Al-Farraj ES, El-Sayed DS, Ahmad R, et al. Experimental and theoretical studies of metal complexes. Scientific Reports. 2024;14:10032. https://doi.org/10.1038/s41598-024-58108-5
CrossRef - Terenti N, Lazarescu A, Shova S, et al. Synthesis, X-ray and antibacterial activity of new copper(II) thiosemicarbazone complexes derived from 4-formyl-3-hydroxy-2-naphthoic acid. Inorganica Chimica Acta. 2024;571:122216. https://doi.org/10.1016/j.ica.2024.122216
CrossRef - Eccles KS, Morrison RE, Maguire AR, Lawrence SE. Crystal landscape of primary aromatic thioamides. Crystal Growth & Design. 2014;14(6):2753–2764. https://doi.org/10.1021/cg401891f
CrossRef - Arshad J, Tong KKH, Movassaghi S, et al. Impact of the metal center and leaving group on the anticancer activity of organometallic complexes of pyridine-2-carbothioamide. Molecules. 2021;26(4):833. https://doi.org/10.3390/molecules26040833
CrossRef - McClendon AK, Osheroff N. DNA topoisomerase II, genotoxicity, and cancer. Mutation Research. 2007;623(1–2):83–97. https://doi.org/10.1016/j.mrfmmm.2007.05.003
CrossRef - Deweese JE, Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Research. 2009;37(3):738–748. https://doi.org/10.1093/nar/gkn937
CrossRef - Wei H, Ruthenburg AJ, Bechis SK, Verdine GL. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. Journal of Biological Chemistry. 2005;280(44):37041–37047. https://doi.org/10.1074/jbc.M506749200
CrossRef - Liang X, Wu Q, Luan S, et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. European Journal of Medicinal Chemistry. 2019;171:129–168. https://doi.org/10.1016/j.ejmech.2019.03.034
CrossRef - Mannaa AH, Mohamed GG, Refaat HM, et al. DFT and molecular docking studies of novel compounds. Scientific Reports. 2025;15:15782. https://doi.org/10.1038/s41598-025-15782-3
CrossRef - Polo-Cuadrado E, Muñoz-Becerra M, Castillo O, et al. Coordination chemistry of transition metals. RSC Advances. 2023;13:30118–30129. https://doi.org/10.1039/d3ra04874h
CrossRef - El-Sayed DS, Al-Farraj ES, Ahmad R, et al. Bioinorganic evaluation of metal complexes. BMC Chemistry. 2025;19:24. https://doi.org/10.1186/s13065-024-01338-5
CrossRef - Mohapatra RK, Das D, Al-Resayes SI, et al. Recent advances in metallodrugs. Frontiers in Pharmacology. 2022;13:1044439. https://doi.org/10.3389/fphar.2022.1044439
CrossRef - Abreu KR, Viana JR, Neto JGO, et al. Exploring thermal stability, vibrational properties, and biological assessments of dichloro(l-histidine)copper(II): a combined theoretical and experimental study. ACS Omega. 2024;9(43):43488–43502. https://doi.org/10.1021/acsomega.4c05029
CrossRef - Balogun TA, Khan SU, Ahmad R, et al. Computational studies of coordination compounds. Frontiers in Chemistry. 2022;10:964446. https://doi.org/10.3389/fchem.2022.964446
CrossRef - Qadri T, Ahmad R, Khan SU, et al. Design and reactivity of metal complexes. RSC Advances. 2023;13:33826–33839. https://doi.org/10.1039/d3ra06149c
CrossRef - Bouloumpasi E, Koskeridou A, Irakli M, et al. Novel copper(II) coordination compounds containing pyridine derivatives of N4-methoxyphenyl-thiosemicarbazones with selective anticancer activity. Molecules. 2024;29(23):5612. https://doi.org/10.3390/molecules29235612
CrossRef - Haribabu J, Panneerselvam M, Swaminathan S, et al. Optimizing nickel complex with thiosemicarbazone for viral applications and cancer therapy. Applied Organometallic Chemistry. 2025;39(9):e70346. https://doi.org/10.1002/aoc.70346
CrossRef - Jyothi P. Recent advances in thiosemicarbazone metal complexes: structure–activity relationship and therapeutic prospects. Discover Chemistry. 2026;3(1). https://doi.org/10.1007/s44371-026-00538-3
CrossRef - Veg E, Hashmi K, Ahmad MI, et al. Biological applications and mechanistic insights of benzaldehyde-substituted thiosemicarbazones and their metal complexes: a review. Natural Sciences. 2025;5(1–2). https://doi.org/10.1002/ntls.70005
CrossRef - Mullaivendhan J, Al-Farraj ES, Ahmad R, et al. Spectroscopic and docking investigations. BMC Chemistry. 2023;17:1067. https://doi.org/10.1186/s13065-023-01067-1
CrossRef - Muğlu H. Synthesis, spectroscopic characterization, DFT studies and antioxidant activity of new 5-substituted isatin/thiosemicarbazones. Journal of Molecular Structure. 2024;1322:140406. https://doi.org/10.1016/j.molstruc.2024.140406
CrossRef - Shiroudi A, Ahmad R, Khan SU, et al. Electronic structure and biological implications. Scientific Reports. 2024;14:58582. https://doi.org/10.1038/s41598-024-58582-x
CrossRef - El-Sherif AA, Mahmoud WH, Abdelkarim AT, et al. Metal complexes featuring a quinazoline Schiff base ligand and glycine: synthesis, characterization, DFT and molecular docking analyses. BMC Chemistry. 2024;18:113. https://doi.org/10.1186/s13065-024-01130-5
CrossRef - Rudrapal M, Khan SU, Ahmad R, et al. Drug discovery approaches using molecular docking. Scientific Reports. 2023;13:35161. https://doi.org/10.1038/s41598-023-35161-0
CrossRef - Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 16, Revision C.01. Wallingford (CT): Gaussian Inc.; 2016.
- Becke AD. Density-functional thermochemistry. III. The role of exact exchange. Journal of Chemical Physics. 1993;98(7):5648–5652. https://doi.org/10.1063/1.464913
CrossRef - Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations: potentials for K to Au including the outermost core orbitals. Journal of Chemical Physics. 1985;82(1):270–283. https://doi.org/10.1063/1.448799
CrossRef - Wadt WR, Hay PJ. Ab initio effective core potentials for molecular calculations: potentials for main group elements Na to Bi. Journal of Chemical Physics. 1985;82(1):284–298. https://doi.org/10.1063/1.448800
CrossRef - Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models. Chemical Reviews. 2005;105(8):2999–3093. https://doi.org/10.1021/cr9904009
CrossRef - Dennington R, Keith T, Millam J. GaussView, Version 6. Shawnee Mission (KS): Semichem Inc.; 2019.
- Kowol CR, Heffeter P, Miklos W, et al. Mechanisms underlying reductant-induced reactive oxygen species formation by anticancer copper(II) compounds. Journal of Biological Inorganic Chemistry. 2011;17(3):409–423. https://doi.org/10.1007/s00775-011-0864-x
CrossRef - Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Research. 2000;28(1):235–242. https://doi.org/10.1093/nar/28.1.235
CrossRef - Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. https://doi.org/10.1002/jcc.21256
CrossRef - Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function. Journal of Computational Chemistry. 2010;31(2):455–461. https://doi.org/10.1002/jcc.21334
CrossRef - Kitchen DB, Decornez H, Furr JR, et al. Docking and scoring in virtual screening for drug discovery. Nature Reviews Drug Discovery. 2004;3(11):935–949. https://doi.org/10.1038/nrd1549
CrossRef - Dassault Systèmes BIOVIA. Discovery Studio Visualizer, Version 2021. San Diego (CA): Dassault Systèmes; 2021.
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 2.5. New York (NY): Schrödinger LLC; 2021.
- Casas JS, García-Tasende MS, Sordo J. Main group metal complexes as drugs. Coordination Chemistry Reviews. 2008;252(14–15):1562–1580. https://doi.org/10.1016/j.ccr.2007.09.009
CrossRef - Hevener KE, Zhao W, Ball DM, et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. Journal of Chemical Information and Modeling. 2009;49(2):444–460. https://doi.org/10.1021/ci800293n
CrossRef

