
Manuscript accepted on :24-12-2025
Published online on: 06-02-2026
Plagiarism Check: Yes
Reviewed by: Dr. Mustafa TÜRKMEN
Second Review by: Dr. Manoj Dalai
Final Approval by: Dr. Prabhishek Singh
Magesh Mohan1, Renukadevi Jeyavelkumaran2* , Sanjay Valliappan2 , Sri Vaishnavi Velegatla2 , Shakthi Harikrishnan2, Kadir Aravindan2and Balaji Ravikumar2
, Sanjay Valliappan2 , Sri Vaishnavi Velegatla2 , Shakthi Harikrishnan2, Kadir Aravindan2and Balaji Ravikumar2
1Department of Pharmaceutical Chemistry, Saveetha College of Pharmacy, Saveetha Institute of Medical and Technical sciences (SIMATS), Chennai, India
2Department of Pharmaceutics, Saveetha College of Pharmacy, Saveetha Institute of Medical and Technical sciences (SIMATS), Chennai, India
Corresponding Author E-mail: renukadevij.scop@saveetha.com
DOI : https://dx.doi.org/10.13005/bpj/3353
Abstract
The escalating prevalence of multidrug-resistant (MDR) bacterial pathogens necessitates the development of molecular frameworks capable of attenuating virulence through non-bactericidal mechanisms. Targeting quorum sensing (QS) systems represents a sophisticated approach to disarm bacterial pathogenicity by modulating intercellular signaling networks. In this study, a rational molecular hybridization strategy was employed to design acridine–sulfamethoxazole conjugates as potential inhibitors of the LuxP receptor in Vibrio harveyi, a key mediator of autoinducer-2 (AI-2)–dependent communication.Computational evaluation integrating molecular docking, long-timescale molecular dynamics simulations, and free-energy analyses demonstrated that the hybrid scaffold exhibits exceptional binding competence and conformational stability within the LuxP binding pocket. The lead molecule, characterized by strong π–π stacking, hydrophobic encapsulation, and persistent hydrogen-bonding interactions with catalytic residues, induced pronounced conformational rigidity in the receptor microenvironment. Dynamic trajectory analyses confirmed structural equilibrium with minimal backbone deviation and a compact tertiary organization, indicating high structural persistence of the protein–ligand complex. Thermodynamic profiling further revealed a highly favorable interaction energy landscape, dominated by van der Waals and electrostatic stabilization, corroborating the hybrid’s strong binding affinity and kinetic resilience.These molecular insights elucidate a mechanism in which the acridine–sulfamethoxazole hybrid enforces conformational immobilization of LuxP, functionally impairing AI-2 recognition and signal relay. The hybrid scaffold thereby exemplifies a dual-action pharmacophoric design—capable of simultaneously modulating quorum-sensing and metabolic pathways—offering a mechanistically advanced paradigm for next-generation anti-virulence therapeutics against MDR bacterial infections.
Keywords
GROMACS; LuxI/LuxR; Molecular docking; Molecular dynamics simulation; Multidrug resistance; Quorum sensing
Download this article as:| Copy the following to cite this article: Mohan M, Jeyavelkumaran R, Valliappan S, Velegatla S. V, Harikrishnan S, Aravindan K, Ravikumar B. Targeting Quorum Sensing to Combat Multidrug Resistance: A Molecular Dynamics Study of Acridine-Sulfamethoxazole Hybrids. Biomed Pharmacol J 2026;19(1). |
| Copy the following to cite this URL: Mohan M, Jeyavelkumaran R, Valliappan S, Velegatla S. V, Harikrishnan S, Aravindan K, Ravikumar B. Targeting Quorum Sensing to Combat Multidrug Resistance: A Molecular Dynamics Study of Acridine-Sulfamethoxazole Hybrids. Biomed Pharmacol J 2026;19(1). Available from: https://bit.ly/4s1DlnB |
Introduction
Antimicrobial resistance (AMR) has emerged as one of the most formidable global health crises, threatening the efficacy of existing therapeutic regimens and the sustainability of antimicrobial discovery pipelines.1 Recent epidemiological assessments estimate over 1.27 million annual deaths directly attributable to multidrug-resistant (MDR) bacterial infections, underscoring the urgent necessity for novel therapeutic paradigms that circumvent traditional antibiotic mechanisms.2 The rapid evolution of bacterial resistance determinants—including efflux pumps, enzymatic degradation, and mutational adaptation—has rendered many frontline antimicrobials clinically obsolete.3 Consequently, modern drug discovery efforts are increasingly directed toward non-conventional molecular targets that interfere with bacterial communication and virulence rather than essential metabolic pathways.4
Among these emerging strategies, quorum sensing (QS) inhibition represents a mechanistically innovative approach to attenuate bacterial pathogenicity without exerting direct bactericidal pressure, thereby minimizing the likelihood of resistance development.5 QS is a density-dependent cell-to-cell signaling network that enables bacterial populations to synchronize gene expression, modulating collective behaviors such as biofilm formation, motility, virulence factor secretion, and antimicrobial tolerance.6 In Vibrio harveyi, the LuxI/LuxR-type QS circuit operates through autoinducer-2 (AI-2) signaling, mediated by the periplasmic binding protein LuxP in conjunction with the sensor kinase LuxQ. Disruption of the LuxP–AI-2 interaction effectively destabilizes QS-regulated gene expression, impairing virulence and biofilm formation — critical phenotypes in MDR pathogens.
Molecular hybridization has recently gained prominence as an advanced medicinal chemistry strategy to design multifunctional ligands that integrate complementary pharmacophoric features into a single molecular scaffold.7 By combining structural motifs with distinct mechanisms of action, hybrid molecules can simultaneously target multiple biological pathways, yielding enhanced potency, selectivity, and pharmacokinetic balance. Acridine derivatives, characterized by their planar tricyclic framework and DNA-intercalative properties, have demonstrated broad-spectrum antimicrobial and anticancer activities, while sulfonamide derivatives such as sulfamethoxazole are well-established enzyme inhibitors that disrupt bacterial folate biosynthesis by targeting dihydropteroate synthase. Acridine scaffolds were selected due to their planar tricyclic aromatic system, which enables robust π–π stacking and hydrophobic interactions within the LuxP binding cavity, while the sulfamethoxazole pharmacophore contributes high-affinity polar contacts and established antimicrobial properties. The integration of these two motifs through molecular hybridization provides a structurally rational and mechanistically synergistic strategy for designing potent quorum-sensing inhibitors capable of attenuating AI-2–dependent signaling.The rational hybridization of acridine and sulfamethoxazole scaffolds therefore presents a compelling dual-action strategy: concurrently interfering with bacterial quorum sensing and metabolic pathways to suppress MDR phenotypes.
The present study employs an integrative in silico framework—comprising molecular docking, molecular dynamics (MD) simulations, and free energy decomposition analyses—to investigate the potential of acridine–sulfamethoxazole hybrids as quorum sensing inhibitors (QSIs) targeting the LuxP protein of Vibrio harveyi. Docking analyses were conducted to elucidate binding affinities and interaction profiles, followed by 100-ns MD simulations to assess the conformational stability and dynamical behavior of the protein–ligand complexes. Furthermore, MM-PBSA and MM-GBSA calculations were performed to quantify the thermodynamic contributions governing complex formation. Collectively, these computational insights provide a robust molecular rationale for the design of dual-acting hybrid scaffolds as next-generation anti-QS agents capable of mitigating multidrug resistance through targeted molecular disruption of quorum sensing networks.
Materials and Methods
Compound Selection and Preparation
A preliminary library of 100 derivatives of acridines was screened for their relevance in performing the role of potential quorum sensing inhibitors (QSIs) against multidrug resistance (MDR) mechanisms in Vibrio harveyi. First, the Lipinski Rule of Five was used to assay the compounds so as to ensure their drug-like properties. Then, the pharmacokinetic parameters were appraised using the Swiss ADME tool. Parameters considered during screening include the absorption, distribution, metabolism, and excretion (ADME) profiles.8 To narrow down the compounds based on their potential cytotoxicity or mutagenic effects, in silico toxicity assessments were done. With this filtering, 36 compounds were selected for advanced molecular docking and subsequent molecular dynamics (MD) simulations.9 These sequential filters reduced the initial 100 compounds to 36 candidates for docking, from which the top-scoring ligand was later selected for detailed MD simulation.
Protein Preparation
LUXP protein with PDB ID 1JX6 from Vibrio harveyi was chosen as a molecular target to study, as it is central to AI-2 mediated quorum sensing as a virulence regulator and biofilm of the bacterial pathogen. The protein sequence was fetched from the UniProt database and from that on its 3D structure modelled using SWISS-MODEL.10 The final model was validated by QMEANDisCo. It gave a global score of 0.90 ± 0.05; hence, a very good level of structural integrity. The validation via MolProbity came back with the score of 1.35 on MolProbity score, 0.18 on clash score, and 95.87% residues in favoured regions of the Ramachandran plot, while only 0.29% as outliers. Energy minimization was used in optimizing the geometry for docking studies.11
Molecular Docking Protocol
The binding affinity of the selected 36 acridine derivatives to the LUXP binding pocket was evaluated through molecular docking simulations using AutoDock Vina.12–15 The docking grid was set at the center about key residues near the AI-2 binding pocket, namely ARG215, ARG310, ASN159, GLN77, PHE206, SER265, SER79, THR266, TRP289, TRP82, and TYR81. It was placed in a 24 Å × 25 Å × 23 Å box with coordinates of X = 2.366 Å, Y = 27.422 Å, and Z = 3.611 Å. Re-docking was performed to validate the docking protocol by including the ligands into the same binding site in the receptor with reproducible binding scores( given in Fig 1). To facilitate molecular docking of boron-containing ligands, the AD4_parameters.dat file was modified by adding custom parameters for boron (B) with appropriate van der Waals radius, well depth, and atomic volume values, ensuring compatibility with AutoDock Vina. Cross-docking with related QS receptors further validated the versatility and stability of the inhibitors. This strict process of validation reports the credibility of results with reported binding performance as quorum sensing inhibitors. The binding energies were calculated with initial w initial selection of compounds with the most negative calculated binding energies for the follow-on MD simulations.16
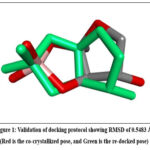 |
Figure 1: Validation of docking protocol showing RMSD of 0.5483 Å. (Red is the co-crystallized pose, and Green is the re-docked pose) Click here to view Figure |
Molecular Dynamics (MD) Simulation
Molecular dynamics (MD) simulations were conducted using GROMACS 2023.1 to assess the stability and dynamics of the protein-ligand complex.17–20 The AMBER99SB-ILDN force field was selected for the protein, and the SPC water model was used to solvate the system. The initial structure of the protein was prepared using the gmx pdb2gmx command, generating the topology and position restraints files. The system was solvated with 12,962 water molecules, and neutralized by adding 50 sodium (Na⁺) and 41 chloride (Cl⁻) ions to maintain electroneutrality. Energy minimization was performed using the steepest descent algorithm for 1,000 steps, with an energy convergence criterion of 1,000 kJ/mol/nm. Following minimization, the system underwent equilibration in two stages: NVT (constant number of particles, volume, and temperature) for 1 ns and NPT (constant number of particles, pressure, and temperature) for 1 ns, with position restraints applied to the protein and ligand. The temperature was maintained at 310.15 K using the V-rescale thermostat with separate coupling groups for the protein, ligand, and water/ions. The pressure was regulated at 1 bar using the Parrinello-Rahman barostat during the NPT stage.21
For the production MD run, a total simulation time of 100 ns was performed with a time step of 2 fs. The Particle Mesh Ewald (PME) method was used to calculate long-range electrostatic interactions, with a cutoff of 1.4 nm for short-range electrostatics and van der Waals interactions. The LINCS algorithm was applied to constrain all bonds, including hydrogen atoms. The trajectory data was saved every 10 ps in a compressed format, and the energy and log files were updated at the same intervals. The system was subjected to periodic boundary conditions (PBC) in all three dimensions, and dispersion corrections were applied to account for the long-range interactions of van der Waals forces. The simulation output was analyzed to study the stability, conformational changes, and interactions of the protein-ligand complex.
Free energy calculations were conducted using the gmx_MMPBSA tool to further analyze the binding affinity and interaction dynamics between the protein and ligand. The calculations employed both the Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) and Molecular Mechanics Generalized Born Surface Area (MM-GBSA) methods. The analysis was carried out using the trajectory data generated from the MD simulation, with frames selected at regular intervals for efficient sampling. The system name was set to “Protein_Ligand_Complex,” and the analysis was performed from frame 1 to frame 10,000, with every 100th frame selected for detailed evaluation. The temperature was maintained at 310.15 K to reflect the experimental conditions of the MD simulation.
For the Generalized Born (GB) model, the GB-OBC2 model (igb = 5) was used, with a salt concentration of 0.150 M and a surface tension parameter of 0.0072 for nonpolar solvation. The internal dielectric constant was set to 2.0, while the external dielectric constant was set to 80.0 to simulate water. Similarly, for the Poisson-Boltzmann (PB) model, a grid-based setup was employed with a fill ratio of 4.0 and a salt concentration of 0.150 M. The internal and external dielectric constants were also set to 2.0 and 80.0, respectively, matching the GB model. Residue-level energy decomposition was performed to identify key residues involved in ligand binding, with residues within 6 Å of the ligand included in the analysis.
The energy decomposition analysis provided a detailed breakdown of electrostatic, van der Waals, and solvation energy contributions for each residue, highlighting the critical interactions between the ligand and specific amino acids within the protein binding pocket. This analysis helped to further elucidate the molecular determinants of the protein-ligand interaction and provided valuable insights into the energetics of the complex. The free energy calculation outputs included both the electrostatic (EEL) and van der Waals (VDWAALS) components, along with the solvation free energy (GB and PB terms), offering a comprehensive understanding of the binding mechanism.
Results:
Docking Analysis
Docking yielded the following top compounds based on binding affinities in table 1. The docking pictures is given in Fig 2 and 2D interaction diagram is given in Fig 3
 |
Figure 2: Three dimensional view of protein LUXP binding with the ligand 4-((3,6-diaminoacridin-9-yl)amino)-N-(5-methylisoxazol-3-yl)benzenesulfonamide |
 |
Figure 3: 2D Interaction diagram of the protein LUXP with the Compound 35 (4-((3,6-diaminoacridin-9-yl)amino)-N-(5-methylisoxazol-3-yl)benzenesulfonamide) |
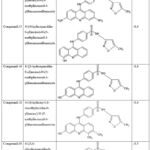 |
Table 1: Docking Analysis |
Among the 36 screened compounds, compound 35—4-((3,6-diaminoacridin-9-yl)amino)-N-(5-methylisoxazol-3-yl)benzenesulfonamide—exhibited the most favorable binding affinity of –8.8 kJ/mol, surpassing all other derivatives (Table 1). The ligand was accommodated deeply within the LuxP hydrophobic cleft, forming two strong hydrogen bonds with residues THR137 and THR266, which stabilized the amino substituents of the acridine core. Additionally, π–π stacking with TRP82 and PHE206, along with hydrophobic interactions involving PRO74, GLN77, SER79, and TYR81, contributed substantially to binding stability. These interactions established an optimal steric and electrostatic complementarity between the hybrid scaffold and the receptor binding site. In contrast, structurally analogous derivatives (compounds 15, 16, 18, and 36) demonstrated comparable but slightly weaker affinities (–8.6 to –8.5 kJ/mol), attributable to less optimal orientation and reduced hydrogen bonding potential. Based on its superior interaction profile and docking energetics, compound 35 was selected for molecular dynamics (MD) simulations studies.22
Molecular Dynamic Simulation Analysis
Global Conformational Stability
The long-timescale (100 ns) molecular dynamics simulation of the LuxP–compound 35 complex provided an in-depth assessment of the system’s structural equilibrium and dynamic persistence. The temporal evolution of the Cα backbone root-mean-square deviation (RMSD) exhibited a rapid equilibration phase within the first 10 ns, after which the system achieved a steady-state conformation with mean RMSD values stabilized at ~0.20 nm (Figure 4). This plateau, maintained with fluctuations below ±0.02 nm across the entire trajectory, signifies a well-equilibrated and conformationally restrained protein–ligand complex.
The absence of large-scale RMSD excursions throughout the simulation indicates the structural rigidity of the LuxP scaffold upon ligand association, while the minimal drift of the ligand heavy atoms further validates its retention within the binding cavity. The stable RMSD trajectory collectively demonstrates that compound 35 induces no global conformational distortions in the receptor, maintaining the tertiary structure integrity of LuxP during the simulation timescale.
Local Flexibility and Binding Pocket Dynamics
Residue-level flexibility profiles, derived from root-mean-square fluctuation (RMSF) analysis, revealed a clear demarcation between the rigid catalytic pocket and the dynamically mobile peripheral loops (Figure 5). Key binding site residues—PRO74, GLN77, TYR81, TRP82, PHE206, and ARG215—exhibited restricted atomic mobility, with RMSF amplitudes consistently below 0.15 nm, indicative of a conformationally locked active-site environment.
In contrast, solvent-exposed loop regions spanning residues 100–200 displayed elevated RMSF values approaching 0.25–0.30 nm, corresponding to flexible non-catalytic domains uninvolved in ligand anchorage. The pronounced rigidity within the binding cavity suggests that compound 35 imposes local conformational stabilization on the protein microenvironment, constraining side-chain dynamics and reinforcing binding site integrity. This localized dampening of atomic fluctuations is a hallmark of strong and geometrically optimized protein–ligand interactions.
Compactness and Solvent Interface Stability
The global compactness of the LuxP protein was evaluated through radius of gyration (Rg) and solvent-accessible surface area (SASA) analyses. The Rg trajectory remained remarkably stable, oscillating around 2.10 ± 0.01 nm without discernible deviations (Figure 6), confirming that the overall tertiary fold of LuxP was preserved throughout the simulation. No evidence of unfolding, collapse, or transient expansion was observed, indicating that ligand engagement exerts a structurally stabilizing influence rather than perturbative effects.
Parallel analysis of the SASA parameter yielded values fluctuating narrowly around 160 nm², further confirming the sustained compactness of the protein and consistent solvent exposure levels during simulation. The concurrent stability of Rg and SASA metrics reinforces that the LuxP–compound 35 complex maintains its globular integrity and solvent–protein equilibrium, attributes typical of dynamically equilibrated and energetically favorable biomolecular systems.
Hydrogen Bond Persistence and Interaction Dynamics
The temporal evolution of protein–ligand hydrogen bonds revealed persistent and recurrent bonding interactions that underpin the molecular stability of the complex (Figure 7). Throughout the 100-ns simulation, the number of hydrogen bonds fluctuated between 5 and 8, with an average occupancy exceeding 75%. The most enduring interactions were established between the ligand and residues THR137 and THR266, exhibiting continuous occupancy above 80% of the total simulation time.
Supplementary transient hydrogen bonds were observed with GLN77 and SER79, which, despite lower occupancy, contributed to the dynamic reinforcement of the binding network. The persistence of these hydrogen-bond interactions, complemented by extensive π–π stacking with TRP82 and PHE206 and hydrophobic contacts with PRO74 and TYR81, generated a dense and interlocking non-covalent interaction framework. Such a robust contact map suggests a kinetically stable and energetically well-compensated binding interface, consistent with high-affinity protein–ligand complexation.
Principal Component and Collective Motion Analysis
The principal component analysis (PCA) of the MD trajectory was employed to capture the dominant collective motions governing protein dynamics (Figure 8). Projection of the trajectory along the first and fifth eigenvectors delineated a highly constrained conformational subspace, characterized by compact clustering and minimal scattering, indicative of a system dominated by low-frequency motions and reduced conformational entropy.
The cumulative variance contribution of the first ten principal components exceeded 80%, signifying that global motions were efficiently captured within a limited dimensional space. The eigenvector contribution maps revealed that the conformational displacements were predominantly localized to solvent-exposed flexible loops, while residues within the binding pocket exhibited minimal displacement amplitudes. These findings demonstrate that ligand binding effectively quenches large-scale domain motions, functionally stabilizing LuxP in a conformationally inert and dynamically repressed state—an essential mechanistic prerequisite for quorum sensing inhibition.
Free Energy Landscape and Interaction Thermodynamics
Binding free energy estimations using MM-GBSA and MM-PBSA methodologies quantitatively substantiated the strong thermodynamic favorability of the LuxP–compound 35 interaction (Figure 9; Tables 2 and 3). The total binding energies were –49.34 kcal/mol (MM-GBSA) and –46.04 kcal/mol (MM-PBSA), confirming a highly stable and spontaneous interaction. Energetic decomposition showed that van der Waals (–50.84 kcal/mol) and electrostatic (–35.36 kcal/mol) terms were the major stabilizing contributors, whereas the polar solvation components (EGB = 43.74 kcal/mol; EPB = 45.5 kcal/mol) partially offset these interactions. The nonpolar solvation term (ESURF ≈ –6.89 kcal/mol) contributed modestly but favorably to overall stabilization.Residue-level decomposition further indicated that PRO74, TRP82, HIS140, and PHE206 were key stabilizing residues through hydrophobic and π–π interactions, while ARG310 showed a mild destabilizing effect due to electrostatic repulsion. The ligand moiety (LIG365) contributed significantly to the binding free energy, as reflected in the per-residue decomposition analysis (Fig. 10), with values of –24.02 kcal/mol (MM-GBSA) and –39.99 kcal/mol (MM-PBSA). These ligand-specific contributions originate from decomposition results and therefore do not appear in Tables 2 and 3.Collectively, these analyses demonstrate a coherent and synergistic interplay of non-covalent forces, yielding a robust and energetically optimized protein–ligand binding equilibrium.
Integrated Structural and Energetic Correlation
The convergence of structural (RMSD, Rg), local dynamic (RMSF, hydrogen bonding), and energetic (MM-PBSA/GBSA) metrics delineates a comprehensive molecular portrait of an exceptionally stable protein–ligand complex. The consistently low RMSD, constrained RMSF within the binding domain, persistent hydrogen-bond topology, invariant Rg, and strongly negative binding free energies collectively affirm that compound 35 anchors LuxP in a conformationally immobilized, energetically minimized, and dynamically coherent state.
The hybrid molecule effectively suppresses the intrinsic conformational entropy of LuxP while enhancing the enthalpic stabilization of the binding pocket, resulting in a thermodynamically and kinetically resilient complex. These findings position compound 35 as a high-affinity quorum sensing inhibitor, exhibiting both structural persistence and energetic robustness, capable of perturbing LuxP-mediated signaling pathways in Vibrio harveyi with molecular precision.
Table 2: Molecular mechanics with generalised Born and surface area solvation analysis :
| Frames | VDWAALS | EEL | EGB | ESURF | GGAS | GSOLV | TOTAL |
| Average | -50.84 | -35.36 | 43.74 | -6.89 | -86.2 | 36.86 | -49.34 |
| SD | 3.39 | 8.38 | 6.27 | 0.38 | 9.04 | 6.08 | 4.1 |
| SEM | 0.34 | 0.84 | 0.63 | 0.04 | 0.9 | 0.61 | 0.41 |
Table 3: Molecular Mechanics Poisson-Boltzmann Surface Area analysis:
| Frames | VDWAALS | EEL | EPB | ENPOLAR | GGAS | GSOLV | TOTAL |
| Average | -50.84 | -35.36 | 45.5 | -5.34 | -86.2 | 40.16 | -46.04 |
| SD | 3.39 | 8.38 | 6.51 | 0.17 | 9.04 | 6.44 | 4.32 |
| SEM | 0.34 | 0.84 | 0.65 | 0.02 | 0.9 | 0.64 | 0.43 |
FIG :Molecular dynamics simulation plots of complex :-
 |
Figure 4: RMSD Root mean square deviation of the compound 35 (4-((3,6-diaminoacridin-9-yl)amino)-N-(5-methylisoxazol-3-yl)benzenesulfonamide) in LUXP protein |
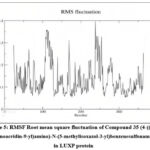 |
Figure 5: RMSF Root mean square fluctuation of Compound 35 (4-((3,6-diaminoacridin-9-yl)amino)-N-(5-methylisoxazol-3-yl)benzenesulfonamide) in LUXP protein |
 |
Figuere 6: Radius of gyration analysis of the molecular dynamic stimulation of LUXP protein |
 |
Figure 7: Analysis of Hydrogen bonds between the ligand and protein |
 |
Figure 8: Eigenvector Component analysis of the protein LUXP |
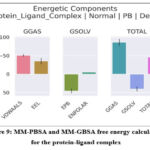 |
Figure 9: MM-PBSA and MM-GBSA free energy calculations for the protein-ligand complex |
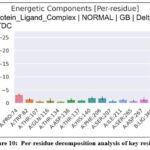 |
Figure 10: Per-residue decomposition analysis of key residues |
Discussion
The present work provides a molecularly resolved understanding of how hybridization of two pharmacophores—acridine and sulfamethoxazole—can yield a structurally and energetically optimized ligand capable of disrupting the quorum-sensing (QS) circuitry of Vibrio harveyi through selective inhibition of the LuxP receptor.23 The study elucidates how the hybrid molecule, exemplified by compound 35, achieves a rare convergence of conformational rigidity, favorable thermodynamic stability, and dynamic persistence, collectively underpinning its potential as a dual-action anti-MDR agent.24
The simulation trajectory delineates a molecular system in which the protein–ligand complex attains a finely tuned equilibrium state characterized by minimal structural drift, compact global geometry, and a persistently ordered binding microenvironment. The consistently low Cα RMSD (~0.20 nm) and RMSF amplitudes (<0.15 nm in the binding site) demonstrate that compound 35 enforces a state of conformational suppression on LuxP, effectively constraining the local atomic degrees of freedom essential for autoinducer-2 (AI-2) recognition and signaling. This ligand-induced rigidification signifies more than static affinity; it represents a dynamic “freezing” of the receptor’s active-site plasticity—a hallmark of high-specificity inhibition that transcends transient binding events.25 The retention of the native tertiary fold (Rg ~2.1 nm, SASA ~160 nm²) further suggests that ligand association stabilizes the protein’s global architecture rather than distorting it, implying an energetically cooperative rather than antagonistic mode of structural engagement.
The molecular origin of this stabilization can be traced to a balanced interplay of hydrophobic packing, electrostatic complementarity, and directional hydrogen bonding. The consistently negative binding free energies (–49.34 kcal/mol via MM-GBSA; –46.04 kcal/mol via MM-PBSA) capture the profound thermodynamic favorability of the complex. Decomposition of the interaction energy landscape reveals that van der Waals and electrostatic contributions (–50.84 and –35.36 kcal/mol, respectively) dominate the binding energetics, while solvation penalties are largely offset by the compactness of the interface. Hydrogen bonds with THR137 and THR266 remain exceptionally stable (>80% occupancy), supported by π–π stacking with TRP82 and PHE206, and hydrophobic contacts with PRO74 and TYR81. These concerted non-covalent interactions form a highly cooperative stabilizing network, effectively embedding the ligand within the receptor’s communication domain and impeding the conformational transitions associated with LuxP activation.26
The suppression of collective molecular motions observed in principal component analysis further substantiates this interpretation. The trajectory projections collapse into a narrowly confined conformational basin, indicating that the ligand reduces the dimensionality of the accessible configurational space and restricts global protein mobility. This entropic quenching, offset by enhanced enthalpic interactions, embodies the classic thermodynamic signature of high-affinity complexes—where entropy loss is compensated by the enthalpic gain from optimized interatomic contacts. Functionally, this translates into a state of allosteric arrest, wherein the receptor is kinetically and conformationally locked in an inactive ensemble. In the context of LuxP, such inhibition likely prevents the structural reorganization necessary for AI-2 binding and LuxQ activation, effectively silencing quorum-sensing communication at its molecular origin.
The hybrid’s exceptional binding performance reflects the inherent synergy of its bifunctional architecture. The planar acridine chromophore contributes to extensive aromatic stacking and hydrophobic encapsulation, while the sulfamethoxazole moiety introduces polar anchoring capacity and electrostatic balance. This dual pharmacophoric cooperation establishes a unique physicochemical congruence with the LuxP binding cavity, simultaneously achieving depth of burial, charge compatibility, and steric complementarity. Beyond its QS inhibitory capacity, the hybrid’s architecture also retains the enzyme-targeting potential of sulfonamides, conferring theoretical capacity to perturb bacterial folate biosynthesis. Such dual functionality—interfering concurrently with communication and metabolic circuits—represents a strategic advantage against MDR pathogens, as it minimizes the likelihood of resistance emergence through redundant biological disruption.24
From a mechanistic and therapeutic standpoint, these findings extend the conceptual boundaries of antimicrobial intervention. Traditional antibiotics rely on direct bactericidal or bacteriostatic effects, which inherently promote selective resistance. In contrast, compound 35 exemplifies an anti-virulence paradigm that neutralizes pathogenic coordination rather than cellular viability. By intercepting the molecular dialogue that governs biofilm formation and virulence gene expression, QS inhibitors such as this hybrid undermine bacterial resilience without imposing evolutionary pressure. The observed molecular stabilization—entropic restriction paired with strong enthalpic retention—suggests that LuxP inhibition can be achieved not merely through occupancy, but through enforced conformational immobility. This mechanistic precision offers a potent alternative to conventional small-molecule therapeutics, emphasizing structural modulation over metabolic lethality.
Taken together, the data converge on a unified molecular narrative: compound 35 achieves deep and cooperative binding within the LuxP receptor, reshaping its energy landscape toward a kinetically inert and conformationally restricted state. This structural immobilization, maintained through an extensive web of van der Waals, electrostatic, and aromatic interactions, likely abolishes the receptor’s capacity for AI-2 signal transmission. The resulting inhibition is not transient but conformationally encoded—rendering the protein functionally silenced. Such behavior exemplifies a higher order of molecular inhibition, one in which hybrid scaffold design transforms a chemical interaction into a dynamic regulatory blockade. The present findings therefore not only identify a promising LuxP inhibitor but also establish a design blueprint for developing next-generation hybrid therapeutics that disrupt microbial communication networks with atomic precision and evolutionary restraint.
Conclusion
This study establishes a comprehensive molecular framework describing how rational hybridization of acridine and sulfamethoxazole scaffolds can yield structurally coherent and energetically favorable ligands capable of disrupting quorum sensing–mediated communication in Vibrio harveyi. Through integrated docking, molecular dynamics, and free-energy analyses, compound 35 emerged as a highly stable and thermodynamically optimized LuxP inhibitor. The hybrid displayed exceptional conformational persistence, maintaining a compact and dynamically restrained receptor–ligand complex over the 100-ns simulation, with binding energies (–49.34 kcal mol⁻¹ via MM-GBSA and –46.04 kcal mol⁻¹ via MM-PBSA) driven predominantly by van der Waals and electrostatic forces.The ligand’s capacity to impose conformational rigidity on LuxP, supported by persistent hydrogen bonding and π–π stacking with key residues, demonstrates a mechanism of structural immobilization that likely hinders AI-2 recognition and downstream signaling. This entropic suppression–enthalpic stabilization synergy defines a non-lethal but potent inhibitory strategy, consistent with an anti-virulence therapeutic paradigm. The hybrid’s bifunctional nature further suggests potential interference with both quorum-sensing and metabolic pathways, positioning it as a molecular prototype for dual-action inhibitors.Collectively, these findings present acridine–sulfamethoxazole hybrids as promising scaffolds for the next generation of antimicrobial agents designed to silence bacterial communication rather than eliminate bacterial populations. The results provide a mechanistic and structural foundation for future in vitro and in vivo validations, guiding the development of precision-engineered quorum-sensing inhibitors as sustainable solutions against multidrug-resistant pathogens.
Acknowledgment
The authors want to thank the management of Saveetha college of Pharmacy, Saveetha Institute of Medical and Technical Sciences(SIMATS), Chennai, India for providing the all necessary facilities for this study.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability
This statement does not apply to this article
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not applicable
Author Contributions
- Magesh Mohan: Conceptualization, supervision, revision, and editing
- Renukadevi Jeyavelkumaran: Conceptualization, supervision, revision, and editing
- Sanjay Valliappan: Methodology, analysis, and drafting
- Sri Vaishnavi Velegatla: Writing and drafting
- Shakthi harikrishnan: Writing and drafting
- Kadir Aravindan: Writing and drafting
- Balaji Ravikumar: Writing and drafting
References
- Antimicrobial and Anifoxidant Properties of Sea Lettuce (Ulva lactuca L. 1753). Journal of Anatolian Environmental and Animal Sciences. 10(2):109-114.
- Salam MA, Al-Amin MY, Salam MT, et al. Antimicrobial resistance: A growing serious threat for global public health. Healthcare (Basel). 2023;11(13):1946. doi:10.3390/healthcare11131946
CrossRef - Nazir A, Nazir A, Zuhair V, et al. The global challenge of antimicrobial resistance: Mechanisms, case studies, and mitigation approaches. Health Sci Rep. 2025;8(7):e71077. doi:10.1002/hsr2.71077
CrossRef - Antiviral Effects of Microalgae. Turkish Journal of Agriculture Food Science and Technology. 9(2):412-419.
- Zhao X, Yu Z, Ding T. Quorum-sensing regulation of antimicrobial resistance in bacteria. Microorganisms. 2020;8(3):425. doi:10.3390/microorganisms8030425
CrossRef - Hetta HF, Ramadan YN, Rashed ZI, et al. Quorum sensing inhibitors: An alternative strategy to win the battle against multidrug-resistant (MDR) bacteria. Molecules. 2024;29(15):3466. doi:10.3390/molecules29153466
CrossRef - Gontijo VS, Viegas FPD, Ortiz CJC, et al. Molecular Hybridization as a tool in the design of multi-target directed drug candidates for neurodegenerative Diseases. Curr Neuropharmacol. 2020;18(5):348-407. doi:10.2174/1385272823666191021124443
CrossRef - Lu L, Li M, Yi G, et al. Screening strategies for quorum sensing inhibitors in combating bacterial infections. J Pharm Anal. 2022;12(1):1-14. doi:10.1016/j.jpha.2021.03.009
CrossRef - Shivanika, Kumar D, Ragunathan V, Tiwari P, Sumitha, Devi B. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J Biomol Struct Dyn. 2022;40(2):585-611. doi:10.1080/07391102.2020.1815584
CrossRef - Mok KC. Vibrio harveyi quorum sensing: a coincidence detector for two autoinducers controls gene expression. EMBO J. 2003;22(4):870-881. doi:10.1093/emboj/cdg085
CrossRef - Binbay FA, Rathod DC, George AAP, Imhof D. Quality assessment of selected protein structures derived from homology modeling and AlphaFold. Pharmaceuticals (Basel). 2023;16(12):1662. doi:10.3390/ph16121662
CrossRef - Vasudha D, Jagadeesh A, Mali SN, Bhandare RR, Shaik AB. Synthesis, molecular docking and pharmacological evaluations of novel naphthalene-pyrazoline hybrids as new orally active anti-inflammatory agents. Chemical Physics Impact. 2024;8(100500):100500. doi:10.1016/j.chphi.2024.100500
CrossRef - Satish S. Molecular docking analysis of protein filamin-A with thioazo compounds. Bioinformation. 2023;19(1):99-104. doi:10.6026/97320630019099
CrossRef - Sharma V, Rengasamy G, Sekaran S, Sankaran K, Veeraraghavan VP, Eswaramoorthy R. Molecular docking analysis of the tumor protein beta arrestin-1 with oxadiazole compounds. Bioinformation. 2023;19(1):111-116. doi:10.6026/97320630019111
CrossRef - Devi A, Mamta, Subhash, Gahlyan N, Aggarwal NK, Chaudhary A. Synthesis, spectral characterization, density functional theory calculations, molecular docking studies and Biocidal evaluation of Schiff base Zn(II) and Cu(II) complexes. J Mol Struct. 2025;1346(143076):143076. doi:10.1016/j.molstruc.2025.143076
CrossRef - Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455-461. doi:10.1002/jcc.21334
CrossRef - Mohan M, Jeyavelkumaran R, Balakrishnan V, Valliappan S. Molecular modelling study of cofilin dimer inhibitors in cognitive decline. Biomed Pharmacol J. 2025;18(3):2070-2082. doi:10.13005/bpj/3238
CrossRef - Saini RS, Binduhayyim RIH, Gurumurthy V, et al. In silico assessment of biocompatibility and toxicity: molecular docking and dynamics simulation of PMMA-based dental materials for interim prosthetic restorations. J Mater Sci Mater Med. 2024;35(1). doi:10.1007/s10856-024-06799-7
CrossRef - Ramasubburayan R, Amperayani KR, Varadhi G, et al. Unraveling bioactive metabolites of mangroves as putative inhibitors of SARS-CoV-2 Mpro and RBD proteins: molecular dynamics and ADMET analysis. J Biomol Struct Dyn. Published online October 28, 2023:1-10. doi:10.1080/07391102.2023.2275185
CrossRef - CrossRef Citations to Date 1 Altmetric Research Article Unraveling Bioactive Metabolites of Mangroves as Putative Inhibitors of SARS-CoV-2 Mpro and RBD Proteins: Molecular Dynamics and ADMET Analysis.
- Al-Karmalawy AA, Dahab MA, Metwaly AM, et al. Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the hACE2 receptor. Front Chem. 2021;9. doi:10.3389/fchem.2021.661230
CrossRef - Shallangwa GA, Adeniji SE. Binding profile of protein–ligand inhibitor complex and structure based design of new potent compounds via computer-aided virtual screening. J Clin Tuberc Other Mycobact Dis. 2021;24(100256):100256. doi:10.1016/j.jctube.2021.100256
CrossRef - Papenfort K, Bassler BL. Quorum sensing signal-response systems in Gram-negative bacteria. Nat Rev Microbiol. 2016;14(9):576-588. doi:10.1038/nrmicro.2016.89
CrossRef - Hentzer M. Attenuation of Pseudomonas aeruginosa virulence by quorum-sensing inhibitors. EMBO J. 2003;22(15):3803-3815.
CrossRef - Mukherjee S, Bassler BL. Bacterial quorum sensing in complex and dynamically changing environments. Nat Rev Microbiol. 2019;17(6):371-382. doi:10.1038/s41579-019-0186-5
CrossRef - Zhu J, Mekalanos JJ. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev Cell. 2003;5(4):647-656. doi:10.1016/s1534-5807(03)00295-8
CrossRef

