
Manuscript accepted on :05-09-2025
Published online on: 22-09-2025
Plagiarism Check: Yes
Reviewed by: Dr. Aybike Turkmen
Second Review by: Dr. Priya Gayathri
Final Approval by: Dr. Mariia Shanaida
Hasanain Abdulhameed Odhar
Department of pharmacology, College of pharmacy, Al-Zahrawi University, Karbala, Iraq.
Corresponding Author E-mail: hodhar3@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/3247
Abstract
Breast cancer is a common type of cancer among females, as it represents 32% of the diagnosed cases in the United States. In the hormone-sensitive kind of breast cancer, the endogenous estrogen is a known promoter for the growth and metastasis of cancerous cells. As age is a major risk factor for the development of breast cancer, this type of cancer is most frequent among postmenopausal women, where estrogen is primarily produced outside the ovaries with the aid of the aromatase enzyme. Therefore, several aromatase inhibitors (AIs) like letrozole and anastrozole were employed for the treatment of estrogen-dependent breast cancer in postmenopausal females. However, the incidence of drug resistance and several side effects are complicating AIs therapy. As such, the objective of this study is to computationally screen a group of phytocompounds against aromatase cytochrome P450 enzyme. Both molecular docking and dynamics simulation tools were used for the structure-based virtual screening to identify new AIs candidates. For this screening, the best results did include three flavonoids (Amentoflavone, Bilobetin, Ginkgetin) and two alkaloids (Irinotecan, Liensinine). Depending on these results, the already approved anticancer chemotherapy Irinotecan is anticipated to have a high drug-likeness score and acceptable pharmacokinetics. Also, docking study pointed to the mostly hydrophobic nature of interactions between Irinotecan and aromatase enzyme. Moreover, the computed binding energy of molecular mechanic-Poisson Boltzmann surface area (MM-PBSA) for Irinotecan during simulation was -10.78 Kcal/ mol and it was one of the best reported values. Finally, the in-vitro anticancer activity of Irinotecan was with micromolar range against susceptible breast cancer cell line.
Keywords
Aromatase cytochrome P450; Docking; Dynamics simulation; Irinotecan; TCM
Download this article as:| Copy the following to cite this article: Odhar H. A. Screening of Traditional Chinese Medicine (TCM) Compounds for their Aromatase Inhibitory Potential by Applying Molecular Docking, Dynamics Simulation and Cytotoxicity Assay. Biomed Pharmacol J 2025;18(3). |
| Copy the following to cite this URL: Odhar H. A. Screening of Traditional Chinese Medicine (TCM) Compounds for their Aromatase Inhibitory Potential by Applying Molecular Docking, Dynamics Simulation and Cytotoxicity Assay. Biomed Pharmacol J 2025;18(3). Available from: https://bit.ly/4gATbku |
Introduction
Breast cancer is currently considered the most commonly diagnosed cancer as it accounts for 32% of all new cases in the United States. Also, breast cancer is the second reason of cancer-related death among the American females with an estimated deaths of 14%.1 It is well-known, that the endogenous estrogens are essential for proliferation and metastasis of breast cancer. In fact, the level of estrogens in breast cancer cells may be up to ten times more than that in plasma of postmenopausal women.2,3 Consequently, the treatment strategy of estrogen-sensitive breast cancer can be directed toward either blocking estrogen receptors or inhibiting estrogen synthesis pathway.4 For the synthesis of endogenous estrogens, the aromatase cytochrome P450 enzyme is mainly involved in the conversion of androgens into estrogens by three successive hydroxylation steps.5
The aromatase enzyme is found as a membrane-bound protein in the endoplasmic reticulum of the estrogen-producing cells. Crystallization of this enzyme showed that the active site of aromatase enzyme has a prosthetic heme group while its binding site contains an androgen-specific cleft. This specificity in binding makes the aromatase enzyme the only vertebrates’ enzyme capable of catalyzing the production of estrogens from androgens.6 Therefore, the inhibition of estrogen production by blocking the aromatase enzyme represents a therapeutic possibility for the treatment of estrogen-dependent breast cancer in postmenopausal women.7 For this group of patients, the third generation aromatase inhibitors (AIs) like letrozole are now approved by the Food and Drug Administration (FDA) as a first-line therapy option.8 However, challenges like treatment resistance and adverse effects like depression and hot flushes are complicating the application of AIs in clinical practice.9 Consequently, the introduction of new AIs molecules that can impede cancer growth with a less incidence of adverse effects represents a current clinical necessity for the treatment of estrogen-sensitive breast cancer in postmenopausal women.10 For this goal, tools for molecular docking and dynamics simulation were used in this study to virtually screen a group of Traditional Chinese Medicine (TCM) compounds against the aromatase enzyme crystal. Then, the best hit compounds were assessed in-vitro for their cytotoxicity against estrogen-sensitive breast cancer cell line. The goal of this in-silico screening study was to find novel phytochemical compounds capable of inhibiting the aromatase enzyme.
Materials and Methods
Setting up the structure-based virtual screening scheme
The major steps of this screening study are schematically clarified in Figure 1. This screening approach was formerly applied in numerous published studies by our lab.11–13 As shown in Figure 1, the initial step of the screening study did involve the docking of TCM compounds library against the active site of the placental aromatase cytochrome P450 enzyme. Then, top five docking hits with the least energy of binding were selected for prediction of their pharmacokinetic, chemical, and toxicity characteristics. Moreover, these selected five hits were submitted to the molecular dynamics (MD) assessment for 50 nanoseconds interval. The results of the MD simulation stage were evaluated through analysis of ligand movement root mean square deviation (RMSD) and binding energy of molecular mechanic-Poisson Boltzmann surface area (MM-PBSA). Finally, these computational findings were further assessed in-vitro by observing the cytotoxicity of the best hit compounds against estrogen-dependent breast cancer cell line.
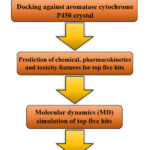 |
Figure 1: A schematic representation for the main stages of the structure-based virtual screening study.Click here to view Figure |
Molecular docking
The DrugRep virtual screen server was utilized for the docking of TCM library of compounds against chain A of the human placental aromatase cytochrome P450.14 Initially, the cytochrome P450 enzyme crystal (PDB: 3EQM) was uploaded to the screening web server.6 It is worth to state that the DrugRep server is employing both AutoDockTools (ADT) ver.1.5.6 and AutoDock Vina ver.1.1.2 to perform the molecular docking process.15,16 During this stage, the applied docking coordinates were X: 84, Y: 50, Z: 50 and the dimensions of the used grid box were 22*22*22 Angstrom. Also, the validity of this docking step was evaluated by redocking the co-crystalized Androstenedione into the active site of cytochrome P450. For this purpose, the PacDOCK tool was used to assess the level of alignment between the docked and co-crystalized Androstenedione.17 After that, the docking hits were arranged based on their predicted energy of binding. For this study, only the best five compounds with minimum energy of binding were chosen for further evaluation. The spatial orientation for each top compound, with a pose of least binding energy, was examined by using both protein-ligand interaction profiler (PLIP) and PyMOL v2.4.1.18 Finally, these selected best five compounds were subjected to the next step for the prediction of various chemical, pharmacokinetics and toxicity properties.
Prediction of pharmacokinetic, chemical, and toxicity properties for the best hits
In this screening step, multiple web-based tools were utilized to predict various pharmacokinetic, chemical, and toxicity features for the best docking hits. The employed webservers include: pkCSM, ProTox 3.0, SwissADME and Molsoft L.L.C.19–21
Molecular dynamics (MD) simulation study
For each of the five top compounds, the ligand-enzyme complex with a pose of least binding energy was submitted to MD simulation for 50 nanoseconds. The YASARA Dynamics v20.12.24 software was used in this study to perform the simulation step.22 The used simulation parameters and procedure in this study are similar to what we have utilized in our former projects.23,24 In summary, the following force fields were used in this MD simulation: TIP3P for H2O, AMBER14 for solute, AM1BCC and GAFF2 for designated ligand.25–27 In addition, during this simulation, a concentration of 0.9% NaCl was implemented to neutralize the ligand-cytochrome P450. The results of this MD simulation were analyzed by considering the ligand proximity to the cytochrome P450 active site as reported by RMSD value throughout 50 nanoseconds interval. Additionally, by using AMBER14 force field, the binding energy for molecular mechanic-Poisson Boltzmann surface area (MM-PBSA) was measured during this simulation.26 For a comparative goal, the Androstenedione-cytochrome P450 docking complex was also submitted as a positive control for the MD simulation process.
Cytotoxicity assay
The MTT test was employed to assess the in-vitro anticancer activity of the top five compounds against the estrogen-sensitive breast cancer cell line (MCF7). The MTT assay was repeated as a duplicate for each compound, the anticancer drug Cisplatin was used as a positive control. Then, the 50% inhibitory concentration (IC50) was calcuated for each tested compound, the final results were reported as mean IC50 ± standard error of mean (SEM) by using GraphPad Prism v7.0. The MTT assay detailed procedure and conditions were similar to what have been described in previous studies.10,28
Results
At first, the accuracy of the employed docking procedure was assessed by the redocking approach. During this approach, the co-crystalized Androstenedione was taken off from the cytochrome P450 chain B crystal. Then, the Androstenedione was docked again to the cytochrome P450 active site by using similar docking procedure. Then, the conformation of the docked Androstenedione was aligned with the co-crystalized one. The variation in the conformation between native and docked Androstenedione was described as RMSD value. This conformations alignment between docked and co-crystalized Androstenedione can be seen in Figure 2. According to this figure, the measured conformations difference RMSD was only 0.47 Angstrom. In addition, the energy of redocking Androstenedione was -13.93 Kcal/ mol.
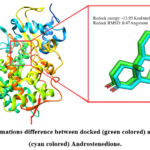 |
Figure 2: Conformations difference between docked (green colored) and co-crystalized (cyan colored) Androstenedione.Click here to view Figure |
After applying the docking step of TCM library against the cytochrome P450 crystal, the best five hits were selected and reported along with their anticipated chemical characteristics in Table 1. These selected top hits were ranked based on their minimum docking energy. As noted from Table 1, all the reported hits are flavonoids except Irinotecan and Liensinine. It is obvious that both Irinotecan and Liensinine are nitrogen-based alkaloids when considering their chemical formula listed in Table 1. Also it is very clear that all the listed hits are violating Lipinski’s rule of five as their molecular weight is higher than 500 g/ mol.29 Moreover, the only hit compound with a logarithm partition coefficient (Log P) greater than 5 is Liensinine. Finally, all the presented hits have a polar surface area (PSA) that exceeds 140 squared Angstrom with the exception of Irinotecan and Liensinine.
Table 1: Chemical characteristics for best five compounds obtained by the docking of Traditional Chinese Medicine (TCM) compounds against the human placental aromatase cytochrome P450 active site. These phytomedicines were listed according to their minimum docking energy.
| No. | Hit name | Chemical formula | Docking score (Kcal/ mol) | M.W. (g/mol) | HBD | HBA | PSA (Å2) | Log P |
| 1 | Amentoflavone | C30H18O10 | -11.8 | 538.5 | 6 | 10 | 181.8 | 3.62 |
| 2 | Bilobetin | C31H20O10 | -11.8 | 552.5 | 5 | 10 | 170.8 | 3.96 |
| 3 | Irinotecan | C33H38N4O6 | -11.6 | 586.7 | 1 | 8 | 114.2 | 3.73 |
| 4 | Ginkgetin | C32H22O10 | -11.4 | 566.5 | 4 | 10 | 159.8 | 4.34 |
| 5 | Liensinine | C37H42N2O6 | -11.3 | 610.7 | 2 | 8 | 83.9 | 5.17 |
M.W.: molecular weight; HBD: hydrogen bond donor group; HBA: hydrogen bond acceptor group; PSA: polar surface area; A: angstrom; Log P: the logarithm of partition coefficient.
Then, various pharmacokinetic and toxicological features were predicted for these best hits and reported in Table 2 along with their drug-likeness score. As observed in Table 2, the only hit compounds with good drug-likeness scores are Irinotecan and Liensinine. Also, all the listed hits are predicted to be hydrophobic compounds except Irinotecan. As such, only Irinotecan is predicted to have a satisfactory volume of distribution and a moderate water solubility. Lastly, all the hits in Table 2 are expected to be not mutagenic. However, only Irinotecan is predicted to has a low median lethal dose (LD50) of less than 1000 mg/ Kg.
Table 2: A prediction overview of drug-likeness score, pharmacokinetic, and toxicity variables for the best screening hits. These phytomedicines were arranged according to their minimum docking energy.
| No. | Compound name | Drug-likeness | Pharmacokinetics | Toxicity | |||
| Water solubility (mg/ml) | Intestinal absorption (%) | VDss(L/Kg) | AMES toxicity | LD50(mg/ Kg) | |||
| 1 | Amentoflavone | 0.19 | 1.36e-06 (poor) | 84.36 | 0.09 | No | 3919 |
| 2 | Bilobetin | 0.27 | 1.10e-06 (poor) | 86.05 | 0.07 | No | 4000 |
| 3 | Irinotecan | 1.54 | 8.67e-04 (moderate) | 99.87 | 13.43 | No | 765 |
| 4 | Ginkgetin | 0.31 | 8.74e-07 (poor) | 95.38 | 0.05 | No | 4000 |
| 5 | Liensinine | 1.55 | 7.13e-06 (poor) | 87.05 | 0.16 | No | 1180 |
VDss: the steady state volume of distribution; LD50: 50% lethal dose.
When considering the selectivity of the top hit compounds against different variants of the cytochrome P450 enzyme in Table 3, it is clear that all these hits have a good selectivity except Liensinine. As can be seen in Table 3, the Liensinine alkaloid is predicted to inhibit several variants of the cytochrome P450 enzyme.
Table 3: Prediction of inhibitory activity for the top five hit compounds against different cytochrome P450 enzyme variants.
| No. | Compound name | Inhibitory activity against cytochrome P450 variants | ||||
| CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | ||
| 1 | Amentoflavone | No | No | No | No | No |
| 2 | Bilobetin | No | No | Yes | No | No |
| 3 | Irinotecan | No | No | No | No | Yes |
| 4 | Ginkgetin | No | No | Yes | No | No |
| 5 | Liensinine | Yes | Yes | No | Yes | Yes |
Moreover, an overview of the herbal sources and the pharmacological effectiveness for the top hits is presented in Table 4. Based on this tabular overview, it is very clear that most of these hit compounds are reported to have anticancer and anti-inflammatory activities.
Table 4: An overview of the botanical sources and the potential activities for best hit compounds in this structure-based virtual screening.
| No. | Hit name | Source | Activity |
| 1 | Amentoflavone | Selaginella tamariscina | Anti-inflammatory, antimicrobial, anticancer.30 |
| 2 | Bilobetin | Ginkgo biloba | Increases the effect of insulin, decreases the level of blood lipids.31 |
| 3 | Irinotecan | Camptotheca acuminata | Anticancer.32 |
| 4 | Ginkgetin | Ginkgo biloba | Anticancer, anti-inflammatory, neuroprotective, antimicrobial.33 |
| 5 | Liensinine | Nelumbo nucifera | Antioxidant, anti-inflammatory, anticancer, antihypertension.34 |
Analysis of the docking complexes, in this screening study, predicted that all the five hit compounds were involved in interactions with several active site amino acid residues of the cytochrome P450 as seen in Figure 3. When considering the compound Androstenedione in Figure 3, it is obvious that this reference ligand can interact with the active site residues similar to those noted with the other hits. Based on these docking results, the main active site residues of the cytochrome P450 that may interact with hit compounds are: Isoleucine 133, Phenylalanine 134, Tryptophane 224, Alanine 306, Aspartic acid 309, Threonine 310, Valine 370, Leucine 372, Valine 373, Methionine 374, Leucine 477 and Serine 478. Also, it is worth to point out that these interactions with the active site residues are mostly hydrophobic in nature. However, this docking study reported few hydrogen bond interactions between hit phytocompounds and the active site amino acids as seen in Figure 3. But these hydrogen bonds were not maintained between ligand and enzyme’s active site when considering dynamic simulation results throughout 50 nanoseconds period.
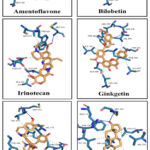 |
Figure 3: Three-dimensional representation for the docking complex of hit compounds and the cytochrome P450 active site, the co-crystalized ligand (Androstenedione) was used as a reference compound. Click here to view Figure |
 |
Figure 4: Hit compounds movement RMSD parameter as a function of simulation duration.Click here to view Figure |
After the analysis of the docking complexes, the five hit compounds together with the reference ligand were submitted to the MD simulation. The results of this simulation are summarized in both Figure 4 and Table 5. Based on this simulation, it is very clear that these five hits were unable to overcome the reference compound (Androstenedione) when considering ligand movement RMSD and MM-PBSA binding energy calculations. As seen in Figure 4 and Table 5, the least average ligand movement was reported for Androstenedione followed by Amentoflavone: 1.36 and 1.70 Angstrom respectively. On the other hand, the best average MM-PBSA binding energy was predicted for the control compound Androstenedione and the hit compound Irinotecan: -3.48 and -10.78 Kcal/mol.
Table 5: A summary for the molecular dynamics (MD) findings of the best hit compounds throughout 50 nanoseconds simulation interval.
| No. | Compound name | MD simulation results | |
| The Mean of ligand movement RMSD (Å) | The average MM-PBSA binding energy (Kcal/ mol) | ||
| 1 | Amentoflavone | 1.70 | -50.43 |
| 2 | Bilobetin | 1.90 | -36.96 |
| 3 | Irinotecan | 3.67 | -10.78 |
| 4 | Ginkgetin | 2.16 | -21.28 |
| 5 | Liensinine | 2.25 | -36.93 |
| 6 | Androstenedione (+ control) | 1.36 | -3.48 |
MD: The Molecular dynamics; RMSD: The Root mean square deviation; Å: Angstrom; MM-PBSA: The Molecular mechanic-Poisson Boltzmann surface area.
Finally, the activity of the five hit compounds against estrogen-dependent breast cancer cell line (MCF7) was determined in-vitro. This potential anticancer activity of these compounds was evaluated by using MTT assay and results were presented in Table 6. Based on Table 6, all the tested compounds were able to achieve a mean IC50 value within micromolar range. Again, none of the five hits was able to exceed the in-vitro anticancer activity reported for the positive control compound (Cisplatin) as observed in Table 6.
Table 6: Summary of MTT assay results.
| No. | Hit compound | Mean IC50 ± SEM (µM) |
| 1 | Amentoflavone | 10.50±1.00 |
| 2 | Bilobetin | 18.00±0.50 |
| 3 | Irinotecan | 22.50±2.00 |
| 4 | Ginkgetin | 31.00±1.50 |
| 5 | Liensinine | 28.00±2.50 |
| 6 | Cisplatin | 1.50±0.50 |
IC50: 50% inhibitory concentration; SEM: Standard error of mean.
Discussion
It is well-known that the endogenous estrogens can stimulate growth and expansion of breast cancer tissue in pre and postmenopausal women. Also, age is regarded a significant risk factor for the breast cancer incidence.35 As such, most of the breast cancer patients are considered postmenopausal women where estrogens are principally produced in the peripheral tissues with the aid of the aromatase enzyme. Consequently, the inhibition of the aromatase cytochrome P450 enzyme constitutes a therapeutic option for the treatment of postmenopausal women with estrogen-sensitive breast cancer.7 However, numerous challenges have emerged against the clinical application of the aromatase inhibitors (AIs) like drug resistance and adverse effects.9 Therefore, the introduction of new AI candidates appears to be a significant clinical priority to overcome these challenges in the management of estrogen-dependent breast cancer. As a result, the objective of this in-silico study is to identify novel inhibitors of the aromatase cytochrome P450 enzyme through screening of TCM library by using tools for molecular docking and dynamics simulation.
Before implementing the structure-based computational screening step, the validity of docking procedure was measured by redocking method. As can be noticed in Figure 2, the conformations alignment RMSD of the docked and native Androstenedione was only 0.47 Angstrom. This very low RMSD value refers to little conformations variation when the docked Androstenedione is aligned with the co-crystalized one. It is known that a good docking accuracy can be inferred when the conformations change RMSD is less than 2 Angstrom.36 In other words, the low conformations variation RMSD of 0.47 Angstrom in this redocking method means that the applied docking procedure has a satisfactory reliability.
Then, the docking step was applied for screening the TCM library of compounds against the aromatase cytochrome P450 chain A. The docking findings were arranged based on the energy of binding for the screened compounds, and the best five hit compounds with least docking energy were selected for further evaluation. At first, several chemical features were predicted for the best five hits as seen in Table 1. Depending on this table, the docking energy was less than -11 Kcal/ mol for all five reported compounds. Also, it is obvious from results in Table 1 that all the five hits are violating Lipinski’s rule of five as their molecular weight is more than 500 g/ mol. Additional violations for rule of five can be noted for Amentoflavone and Liensinine when considering the number of hydrogen bond donor groups (HBD >5) and the logarithm of partition coefficient (Log P >5) respectively. All these predicted violations for rule of five may adversely influence the oral bioavailability of these phytocompounds.29 Lastly, all the phytocompounds in Table 1 are expected to have a high polar surface area (PSA) of more than 140 Å2 except for Irinotecan and Liensinine. This anticipated high PSA value may further contribute to poor oral bioavailability of these compounds by violating Veber’s rule.37
In addition, different pharmacokinetics and toxicity characteristics were anticipated for the top five hits together with their drug-likeness score as reported in Table 2. As seen in this table, only the compounds Irinotecan and Liensinine are expected to have a high drug-likeness score. Moreover, only the compound Irinotecan is believed to have a moderate water solubility and a subsequent good steady state volume of distribution (VDss). As such, only Irinotecan is believed to have acceptable pharmacokinetics. Although all the listed compounds in Table 2 are anticipated to be non-mutagenic, but Irinotecan is reported to have a low median lethal dose (LD50) of less than 1000 mg/Kg which may raise some safety concerns regarding this compound.
One of the challenges that can face the clinical application of AIs is the possibility of having serious adverse effects due to the nonselective inhibition of cytochrome P450 several variants. The nonselective inhibition of the cytochrome P450 can interfere with the endogenous biosynthesis of cortisol and aldosterone leading to these adverse effects.10 For this reason, the five hit compounds were then assessed for their potential activity against several variants of the cytochrome P450 enzyme as seen in Table 3. Based on this table, only the last hit Liensinine is believed to have a non-selective activity against several variants of the enzyme.
When reviewing previous literatures for the herbal source and the potential pharmacological activity of the five hit compounds, it is quite clear that many of these hits are believed to have anticancer and anti-inflammatory effects as listed in Table 4. Of interest is the alkaloid Irinotecan which is a known topoisomerase inhibitor that is already approved for treatment of colon and lung cancer.32 As such, the compound Irinotecan may represent a potential candidate that can be repurposed as an aromatase cytochrome P450 inhibitor and then it can be employed in the management of estrogen-sensitive breast cancer.
Crystallization studies showed that the aromatase cytochrome P450 is considered an androgen specific enzyme. The enzyme active site contains multiple of hydrophobic and polar residues that allow the entrance of lipophilic substrates.6 The examination of docking images in Figure 3 points to the possibility that the five hit compounds are interacting with similar enzyme active site residues as does the reference compound Androstenedione. These docking interactions are mostly hydrophobic in nature with few hydrogen bonds. However, the predicted hydrogen bonds were not maintained when each ligand-target docking complex was subjected to the MD simulation.
A summary for the MD simulation findings is shown in both Table 5 and Figure 4. In this simulation, the ligand closeness to the enzyme active site was reported as mean ligand movement RMSD. It is well-known that a low ligand movement RMSD usually refers to the possibility of strong interaction between ligand and target.38 As noted from Table 5, the best mean ligand movement RMSD value was measured for the reference Androstenedione and the hit Amentoflavone: 1.36 and 1.70 Å respectively. While the least proximity to active site was measured for Irinotecan with a mean ligand movement RMSD value of 3.67 Å. A detailed plot for the ligand movement RMSD value of each compound can be noticed in Figure 4 during simulation interval. The strength of interaction between ligand and target can be also interpreted during simulation by considering the average MM-PBSA binding energy. In this case, a more positive binding energy usually refers to superior interaction between ligand and target.11,12 In this regard, it is expected that both Androstenedione and Irinotecan do have the best average MM-PBSA binding energy of -3.48 and -10.78 Kcal/ mol respectively as mention in Table 5.
Lastly, the in-vitro anticancer activity was tested for the five hit compounds along with the positive control Cisplatin. For this purpose, the MTT assay was used to measure the effect of these compounds against estrogen-sensitive breast cancer (MCF7 cell line). As can be noted in Table 6, all the hit compounds have a considerable IC50 inhibitory concentration within micromolar range. However, none of the tested compounds was able to exceed the inhibitory activity produced by the control compound Cisplatin.
Conclusion
In this in-silico study, compounds in the Traditional Chinese Medicine (TCM) library were screened against the aromatase cytochrome P450 enzyme by using docking and molecular dynamics simulation. As a result of this screening, three flavonoids (Amentoflavone, Bilobetin, Ginkgetin) and two alkaloids (Irinotecan, Liensinine) were identified as potential inhibitors of the enzyme active site. Interestingly, the alkaloid Irinotecan is an approved drug for the treatment of colon and lung cancer. According to the prediction analysis, this alkaloid has a high drug-likeness score, moderate water solubility, good volume of distribution and high intestinal absorption. Moreover, Irinotecan seemed to be selective in inhibiting the aromatase cytochrome P450 through multiple hydrophobic interactions and few hydrogen bonds. During the molecular dynamics (MD) simulation, the alkaloid Irinotecan was only able to maintain a mean ligand movement RMSD value of 3.67 Å but the calculated average MM-PBSA binding energy value for this compound was among the best of -10.78 Kcal/ mol. Finally, the in-vitro MTT assay demonstrated that this alkaloid has a mean IC50 inhibitory concentration within micromolar range against estrogen-dependent breast cancer cell line. Based on these findings, it is believed that the alkaloid Irinotecan may represent a potential candidate for repurposing as an aromatase inhibitor in estrogen-sensitive breast cancer patients. However, these preliminary findings have to be confirmed by in-vitro and in-vivo evaluation.
Acknowledgment
The author would like to thanks the college of pharmacy, Al-Zahrawi University for their support of this work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Authors’ Contribution
The sole author was responsible for the conceptualization, methodology, data collection, analysis, writing, and final approval of the manuscript.
References
- Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025. CA Cancer J Clin. 2025;75(1):10.
CrossRef - van Landeghem AA, Poortman J, Nabuurs M, Thijssen JH. Endogenous concentration and subcellular distribution of estrogens in normal and malignant human breast tissue. Cancer Res. 1985;45(6):2900-2906.
- Russo J, Lareef MH, Balogh G, Guo S, Russo IH. Estrogen and its metabolites are carcinogenic agents in human breast epithelial cells. J Steroid Biochem Mol Biol. 2003;87(1):1-25.
CrossRef - Suvannang N, Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V. Molecular Docking of Aromatase Inhibitors. Molecules. 2011;16(5):3597.
CrossRef - Chan HJ, Petrossian K, Chen S. Structural and Functional Characterization of Aromatase, Estrogen Receptor, and Their Genes in Endocrine-Responsive and – Resistant Breast Cancer Cells. J Steroid Biochem Mol Biol. 2015;161:73.
CrossRef - Ghosh D, Griswold J, Erman M, Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nat 2009 4577226. 2009;457(7226):219-223.
CrossRef - Chumsri S, Howes T, Bao T, Sabnis G, Brodie A. Aromatase, aromatase inhibitors, and breast cancer. J Steroid Biochem Mol Biol. 2011;125(1-2):13-22.
CrossRef - Riemsma R, Forbes CA, Kessels A, et al. Systematic review of aromatase inhibitors in the first-line treatment for hormone sensitive advanced or metastatic breast cancer. Breast Cancer Res Treat. 2010;123(1):9-24.
CrossRef - Sledge GW, Mamounas EP, Hortobagyi GN, Burstein HJ, Goodwin PJ, Wolff AC. Past, present, and future challenges in breast cancer treatment. J Clin Oncol. 2014;32(19):1979-1986.
CrossRef - Acar Çevik U, Kaya Çavuşoğlu B, Sağlık BN, et al. Synthesis, Docking Studies and Biological Activity of New Benzimidazole- Triazolothiadiazine Derivatives as Aromatase Inhibitor. Mol 2020, Vol 25, Page 1642. 2020;25(7):1642.
CrossRef - Odhar HA, Hashim AF, Ahjel SW, Humadi SS. VIRTUAL SCREENING OF FDA-APPROVED DRUGS BY MOLECULAR DOCKING AND DYNAMICS SIMULATION TO RECOGNIZE POTENTIAL INHIBITORS AGAINST MYCOBACTERIUM TUBERCULOSIS ENOYL-ACYL CARRIER PROTEIN REDUCTASE ENZYME. Int J Appl Pharm. 2024;16(1):261-266.
CrossRef - Odhar HA, Hashim AF, Humadi SS, Ahjel SW. LIGAND-BASED VIRTUAL SCREENING OF FDA-APPROVED DRUGS TO IDENTIFY NEW INHIBITORS AGAINST LACTATE DEHYDROGENASE ENZYME OF MALARIA PARASITES. Int J Appl Pharm. 2024;16(1):255-260.
CrossRef - Odhar HA, Odhar ZA, Muhi MR. Screening of Traditional Chinese Medicine Library Against Penicillin-binding Protein 2a for Methicillin-resistant Staphylococcus aureus by Molecular Docking, Dynamics Simulation and In vitro Antimicrobial Activity. Biomed Pharmacol J. 2025;18(1):823-834.
CrossRef - Gan J hong, Liu J xiang, Liu Y, et al. DrugRep: an automatic virtual screening server for drug repurposing. Acta Pharmacol Sin. 2023;44(4):888-896.
CrossRef - Morris GM, Ruth H, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30(16):2785.
CrossRef - Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J Chem Inf Model. 2021;61(8):3891-3898.
CrossRef - Carbone J, Ghidini A, Romano A, Gentilucci L, Musiani F. PacDOCK: A Web Server for Positional Distance-Based and Interaction-Based Analysis of Docking Results. Molecules. 2022;27(20):6884.
CrossRef - Schake P, Bolz SN, Linnemann K, Schroeder M. PLIP 2025: introducing protein-protein interactions to the protein-ligand interaction profiler. Nucleic Acids Res. 2025;53(W1).
CrossRef - Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem. 2015;58(9):4066.
CrossRef - Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7.
CrossRef - Banerjee P, Kemmler E, Dunkel M, Preissner R. ProTox 3.0: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024;52(W1):W513-W520.
CrossRef - Krieger E, Vriend G. YASARA View – molecular graphics for all devices – from smartphones to workstations. Bioinformatics. 2014;30(20):2981-2982.
CrossRef - Odha HA, Alhaideri MRA, Hussein RM, Al-Rahmany HA, Hussein SF, Geldenhuys WJ. Computer aided design and characterization of novel acetylcholinesterase enzyme inhibitor for potential utilize in Alzheimer’s disease therapy. Indian J Forensic Med Toxicol. 2019;13(4):804-809.
CrossRef - Odhar HA, Kareem SM, Alhaideri MRA, Hasan MA, Geldenhuys WJ. The design and evaluation of a novel monoamine oxidase b inhibitor through in silico approach. Int J Pharm Qual Assur. 2019;10(2):349-355.
CrossRef - Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696-3713.
CrossRef - Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general Amber force field. J Comput Chem. 2004;25(9):1157-1174.
CrossRef - Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23(16):1623-1641.
CrossRef - Çevik UA, Sağlık BN, Ardıç CM, Özkay Y, Atlı O. Synthesis and evaluation of new benzimidazole derivatives with hydrazone moiety as anticancer agents. Turkish J Biochem. 2018;43(2):151-158.
CrossRef - Lipinski CA. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv Drug Deliv Rev. 2016;101:34-41.
CrossRef - Xiong X, Tang N, Lai X, et al. Insights Into Amentoflavone: A Natural Multifunctional Biflavonoid. Front Pharmacol. 2021;12:768708.
CrossRef - Kou XH, Zhu MF, Chen D, et al. Bilobetin ameliorates insulin resistance by PKA-mediated phosphorylation of PPARα in rats fed a high-fat diet. Br J Pharmacol. 2012;165(8):2692.
CrossRef - Bailly C. Irinotecan: 25 years of cancer treatment. Pharmacol Res. 2019;148.
CrossRef - Adnan M, Rasul A, Hussain G, et al. Ginkgetin: A natural biflavone with versatile pharmacological activities. Food Chem Toxicol. 2020;145.
CrossRef - Zhou J, Li G, Zheng Y, et al. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy. 2015;11(8):1259.
CrossRef - Łukasiewicz S, Czeczelewski M, Forma A, Baj J, Sitarz R, Stanisławek A. Breast Cancer—Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers (Basel). 2021;13(17):4287.
CrossRef - Hevener KE, Zhao W, Ball DM, et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model. 2009;49(2):444-460.
CrossRef - Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615-2623.
CrossRef - Odhar HA, Ibrahim AA, Hashim AF. Computational Screening of Traditional Chinese Medicine (TCM) Library to Identify Potential Inhibitors of H5N1 Avian Influenza Neuraminidase: A Molecular Docking and Dynamics Simulation Study. Biomed Pharmacol J. 2025;18(2):1611-1621.
CrossRef
List of Abbreviations
Å: Angstrom.
AI: Aromatase inhibitor.
FDA: Food and drug administration.
IC50: 50% inhibitory concentration.
LD50: Median lethal dose.
Log P: Logarithm partition coefficient.
MD: Molecular dynamics.
MM-PBSA: Molecular mechanics-Poisson Boltzmann surface area.
PSA: Polar surface area.
RMSD: Root mean square deviation.
SEM: Standard error of mean.
TCM: Traditional Chinese medicine.
VDss: steady state volume of distribution.

